
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Understanding Interchangeable Biosimilars at the Federal and State Levels
ABSTRACT
Though biologic medications have improved the treatment landscape by providing the solution to many unmet medical needs, biologics cost much
more than small-molecule chemical medications. Biosimilars are cost-effective alternatives that are highly similar to originator/reference biologics in terms of efficacy and safety, yet the uptake of biosimilars in the United States has been gradual in part due to provider- and patient-related barriers. To improve biosimilar uptake and adoption, the FDA has established an interchangeable designation for biosimilars. This review discusses the FDA biosimilar and interchangeable biosimilar approval pathway requirements, FDA regulations, and state-specific requirements regarding the dispensing and administration of biosimilars and interchangeable biosimilars. For FDA approval, a biosimilar must demonstrate that it is highly similar to the reference product and that there are no clinically meaningful differences between the reference biologic and the proposed biosimilar. Interchangeable biosimilars are recommended to undergo additional studies to demonstrate that there is no change in efficacy or safety after 3 switches between the proposed interchangeable biosimilar and the reference product. Most states permit the substitution of a reference product for an interchangeable biosimilar without the approval of the prescribing physician. The time frame required for communication to the prescriber about such substitutions varies by state. This review highlights the importance of understanding the regulation of interchangeable biosimilar products in each state.
https://doi.org/10.37765/ajmc.2023.89419
Introduction
Biologic drugs are at the forefront of medicine. They have tremendous benefit for patients with historically unmet medical needs, and they enable improved development of precision medicine. With numerous molecules in


Humphreys

the preclinical or clinical trial phase of development, biologics have the potential to solve the unmet treatment need for many diseases and conditions, such as multiple sclerosis, Crohn disease, asthma, psoriatic arthritis, rheumatoid arthritis, chronic pain, and cancer.1 For example, a biologic medication produced in the living cells of patients with cancer produces a chimeric antigen receptor (CAR) specific for the tumor antigen during CAR T-cell therapy.2 The biopharmaceutical industry has stepped up its already robust efforts to develop new and better biologics to address a multitude of still-unmet needs.
In addition to new drug development, the industry has also focused on providing economically accessible biologics in the form of biosimilars. As a result, biosimilar use is becoming more common. In the United States, biosimilar use in oncology increased up to 80%, depending on the biosimilar, from 2019 to 2021.3 The increased uptake of biosimilars has already resulted in substantial savings to the US health care system; additional savings of approximately $181 billion during the next 5 years is anticipated as new biosimilars launch and adoption of existing biosimilars increases.3,4
As biologics and their more affordable biosimilar versions become increasingly common in prescribing practices across therapeutic areas, health care providers need an enhanced understanding of these drugs. Awareness of the differences in regulations between biosimilars, biologics, and small-molecule medications is key. This becomes increasingly complex when considering interchangeability designations. Thus, this review describes the differences in how the FDA approves, regulates, and permits substitutions for biosimilars versus reference biologic products, with a particular focus on interchangeable biosimilars. This article also highlights the importance of understanding the regulation of interchangeable biosimilar products by state in the United States.
Biologic vs Small-Molecule Medicines
Brand-name chemical drugs, or innovator compounds, are chemically synthesized small molecules.5 Identically replicating these drugs is generally straightforward, due to their small size and simple structure. For example, aspirin is easily synthesized because it is made of chemicals and is comprised of only 21 atoms.1 Generic products, which contain the identical compound to the brand-name drug, provide lower-cost alternatives to the expensive innovator compounds.5
Biologic drugs are different. Biologics are produced in a living system (such as microorganisms or animal cells) and are typically large, complex molecules.6 Typically, biologics display certain levels of heterogeneity, mostly due to posttranslational modifications during the manufacturing process of biologics in living organisms. In addition, inherent differences between manufactured lots are normal and expected.7,8 Biologics are much larger molecules compared with those that have been chemically synthesized.5 For example, a monoclonal antibody such as an immunoglobulin G antibody comprises approximately 25,000 atoms, which is more than 1000 times larger than an aspirin molecule.1 Thus, it is impossible to manufacture a completely identical protein to a reference biologic. Furthermore, because of the complicated development and manufacturing processes, these biologic medications are marketed at a much higher cost than the small-molecule chemical medications.9 These costs puts additional strain on the industry, which already has been stretched thin by the 3-year fight against COVID-19.
Biosimilars are cost-effective alternatives to biologics that are highly similar to reference biologics in terms of efficacy, safety, and immunogenicity, without clinically meaningful differences.6,9 Just as there are certain levels of heterogeneity within a lot and potential small variations between lots of the same biologic, a biosimilar might have minor differences when compared with the reference biologic. These differences are due to the complicated large protein molecule structures, posttranslational modifications, and the manufacturing process.8,10
Biologic and Biosimilar Approval Pathways
A biologic product is approved through a different pathway than a biosimilar. A reference biologic will gain approval via the 351(a) pathway, whereas the approval of a biosimilar occurs via the abbreviated 351(k) pathway.11 When a new biologic medicine is seeking FDA approval for marketing, a standalone approval is required.11 This means that the manufacturer sponsoring the application must conduct sufficient clinical trials, including phase 3 trials, to prove efficacy, safety, tolerability, and purity.9 The goal is to independently demonstrate efficacy and safety for the full profile of the medication.9
The abbreviated approval pathway, 351(k), is different from the standalone pathway, 351(a).11 The abbreviated approval pathway was established under the Patient Protection and Affordable Care Act; within the Affordable Care Act is the Biologics Price Competition and Innovation Act (BPCIA).12 BPCIA has many similarities to the Drug Price Competition and Patent Term Restoration Act (1984), which is also known as the Hatch-Waxman Act. The latter outlines the abbreviated new drug application that pharmaceutical manufacturers may submit for FDA approval of a generic drug. Importantly, the Hatch-Waxman Act applies only to small molecules (ie, molecules manufactured through chemical synthesis), and it requires generic products to be bioequivalent to their brand-name counterparts.13 However, because of the structural complexity and heterogeneity inherent of biologics, it is not possible for an identical copy of a biologic medication to be made.8 Therefore, it is impossible to produce generic versions of high-cost biologic medications; hence the need for biosimilars.
The abbreviated approval pathway for biosimilars, under BPCIA, focuses on the totality of the evidence for the biosimilar. This evidence includes, but is not limited to, the functional and structural attributes, pharmacokinetics, pharmacodynamics, immunogenicity, efficacy, and safety of the proposed biosimilar product.12 To gain approval, a biosimilar product needs to be highly similar (not identical) to a US-licensed reference product, with no clinically meaningful differences to the reference product.7
The FDA does not require the proposed biosimilars be investigated for all indications for which the reference product is licensed, granted that there is sufficient scientific justification.12 This is called indication extrapolation. This concept helps shorten the biosimilar development and approval process, which reduces the overall cost of biosimilar research and development. Biosimilars can then be marketed at reduced costs to patients and health care systems.14
FDA Regulation of Biosimilars
FDA regulations for biosimilars and biologics are different than the regulations for small-molecule generic products. The FDA permits pharmacists to substitute any brand-name small-molecule product for the generic version, provided that the prescriber does not specify “Dispense as written” on the script.15 In contrast, pharmacists are not permitted to dispense a biosimilar product if the reference product of a biologic has been prescribed.15 When a patient is prescribed a reference biologic, the FDA requires authorization from the prescriber before a biosimilar can be dispensed.15 This is because, although highly similar, biosimilars are not completely identical to the reference product, and therefore they cannot be substituted in the same way generic products can.
Interchangeable Biosimilars
The interchangeable designation, established by BPCIA, presents an alternative to the aforementioned rule prohibiting biosimilar substitution. The interchangeable designation allows an interchangeable biosimilar to be automatically substituted for the original reference biologic, subject to state pharmacy laws.6 For a biosimilar to be considered interchangeable, it must satisfy 3 conditions. First, it must be an FDA-approved biosimilar; second, it must produce the same clinical outcome as the reference product in any given patient for any given indications; and third, if the biosimilar medication is given more than once to the same patient, either switching from the reference product or alternating with the reference product, it must produce the same efficacy and have no greater risk of adverse events as when the reference product is used alone (with no switching or alternating).16
All states have pharmacy laws that employ a definition of an interchangeable biosimilar and provide laws for interchangeable biosimilar substitutions. However, these provisions vary from state to state. Pharmacists must take caution in noting these variations. US policy is different from that in the European Union (EU). The European Medicines Agency and Heads of Medicines Agencies allow all biosimilars that are approved in the EU to be used interchangeably with their reference products and other biosimilars equivalent to the same reference product. There are no additional requirements and separate designation for interchangeable biosimilars.17
To support companies designing clinical studies to obtain interchangeability designation, the FDA has a guidance document titled “Considerations in Demonstrating Interchangeability With a Reference Product Guidance for Industry.”16 There is an ongoing discussion among clinicians regarding the necessity of an interchangeability designation in the United States, with some sharing the view that the additional demand of efficacy and safety data goes beyond the evidence required for biosimilar approval.18,19
Regardless, although not required, the FDA document suggests a study investigating 3 switches between the reference product and the proposed interchangeable biosimilar.16 The FDA is flexible about the approach the applicant takes because it may depend on the complexity of the reference product and the postmarket information for the reference product and the biosimilar (if applicable). However, there are some general factors that are suggested for consideration when designing a switching study to establish interchangeability.16 Generally, the study should have a lead-in period where all patients are treated with the reference product and then randomly assigned to 1 of 2 arms: one with multiple switches between the proposed interchangeable biosimilar and the reference product (the switching arm), and the other with continued reference product treatment (nonswitching arm) (Figure 1.)16


Figure 1.
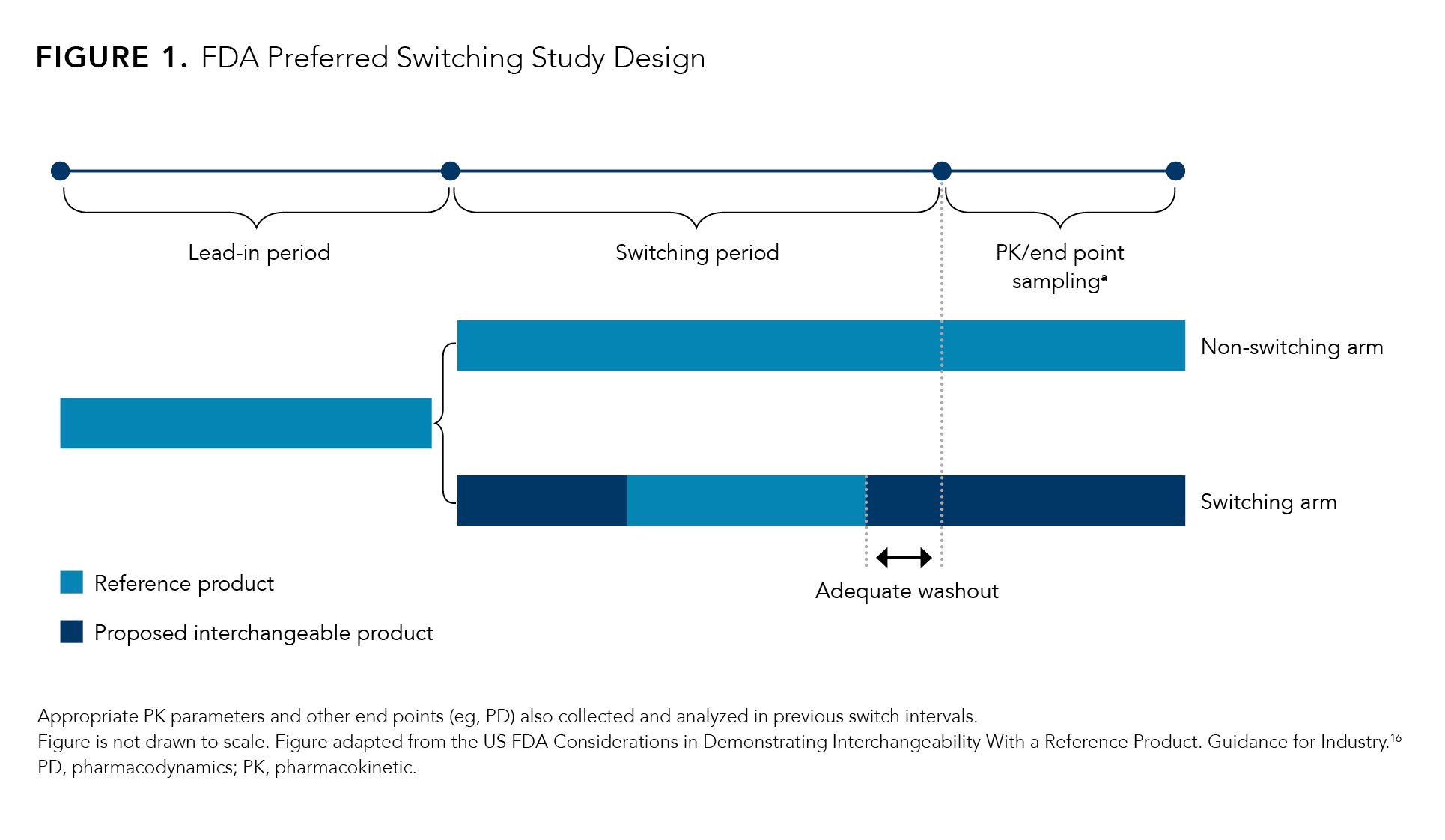
The end points of these switching studies should assess clinical response in terms of safety and diminished efficacy through an assessment of the potential differences in immunogenicity, pharmacokinetics and/or pharmacodynamics for patients in both the switching and nonswitching arms.16 Dropout rates for both arms must be disclosed, and any observed differences must be explained.16 The number of switches and the duration of each treatment period may vary depending on the indication(s) being investigated.16
In addition, the FDA requires robust postmarket safety monitoring for the interchangeable biosimilar.16 A biosimilar product that receives the first interchangeable designation has exclusivity for a full year. It is possible that multiple interchangeable biosimilars could share a period of initial interchangeable exclusivity if they are approved on the same day.20
Although the FDA does not require that interchangeable biosimilars seek licensures for all indications for which the reference product is licensed, it is recommended.16 Thus, in most cases, the interchangeable biosimilar would have the same indications as the reference product. This provides assurance to patients and providers that a patient can be switched from a reference product to the interchangeable biosimilar product. Switching studies can help ensure the high quality of the interchangeable biosimilar products in terms of efficacy, safety, and immunogenicity, as well as similarity with the reference product.16
FDA Regulations for Interchangeable Biosimilars by State
The FDA allows pharmacy-level substitution of a reference product with an interchangeable biosimilar without the prescribing physician’s authorization. However, the regulations regarding automatic substitution from a reference product to an interchangeable biosimilar vary among states. Because state regulations vary, it is crucial that pharmacists adhere to the state-specific laws and regulations.21 Arkansas, Idaho, Kansas, Maine, Mississippi, New Jersey, North Carolina, Ohio, and Vermont permit substitution of a reference product to an interchangeable biosimilar only if the cost to patients for the interchangeable biosimilar product is lower than the reference product.21 In total, 47 of the 50 states allow pharmacist substitution of the prescribed reference product to an interchangeable biosimilar without the authorization of the physician, provided that this is communicated back to the prescribing physician and/or patient. Only 4 states, Alabama, Indiana, South Carolina, and Washington, do not allow automatic substitution at the pharmacy level (Figure 2).


Figure 2.
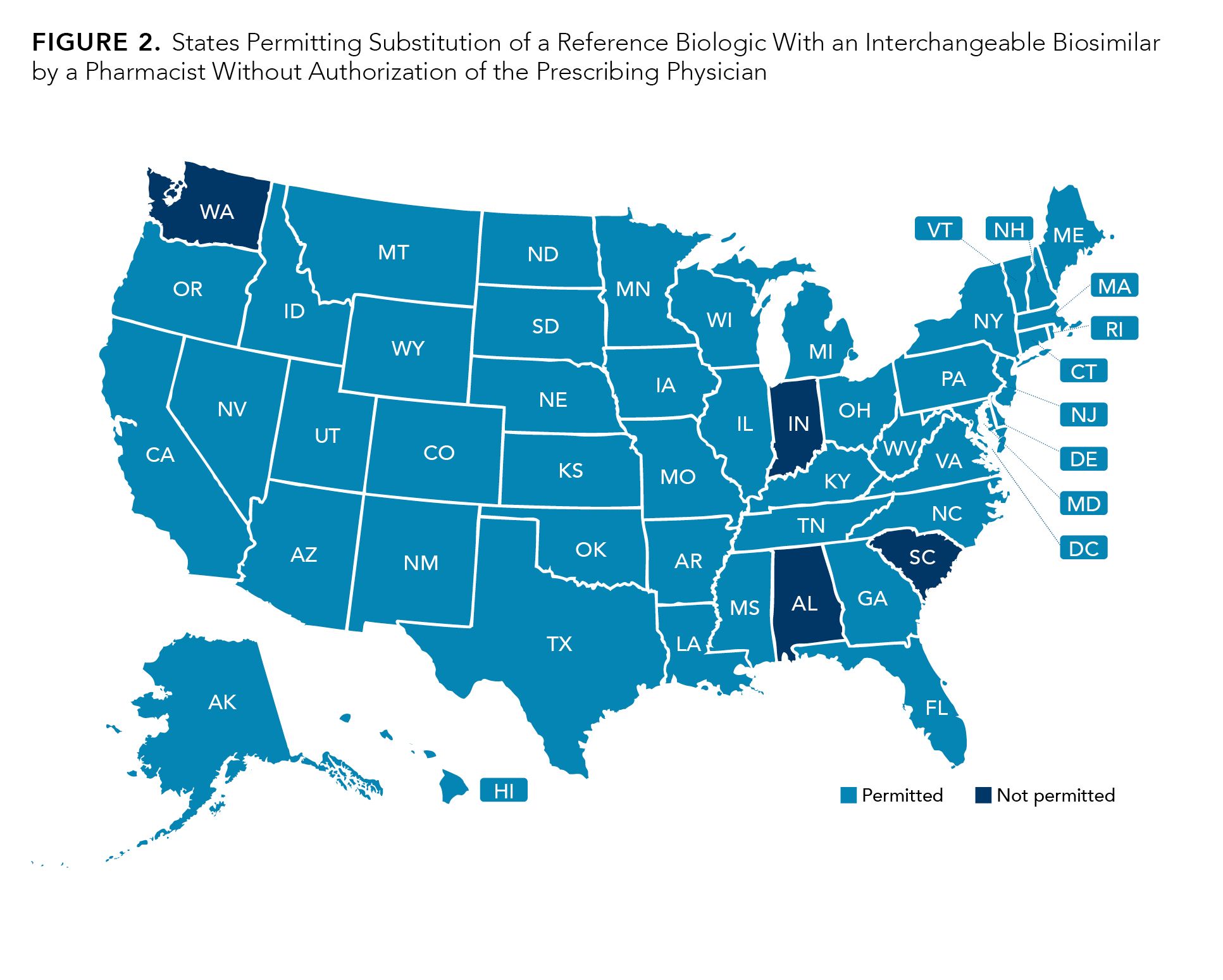
It is important to note that prescribers have the authority to prevent automatic substitution of the reference product to an interchangeable product by requesting “dispense as written.”21
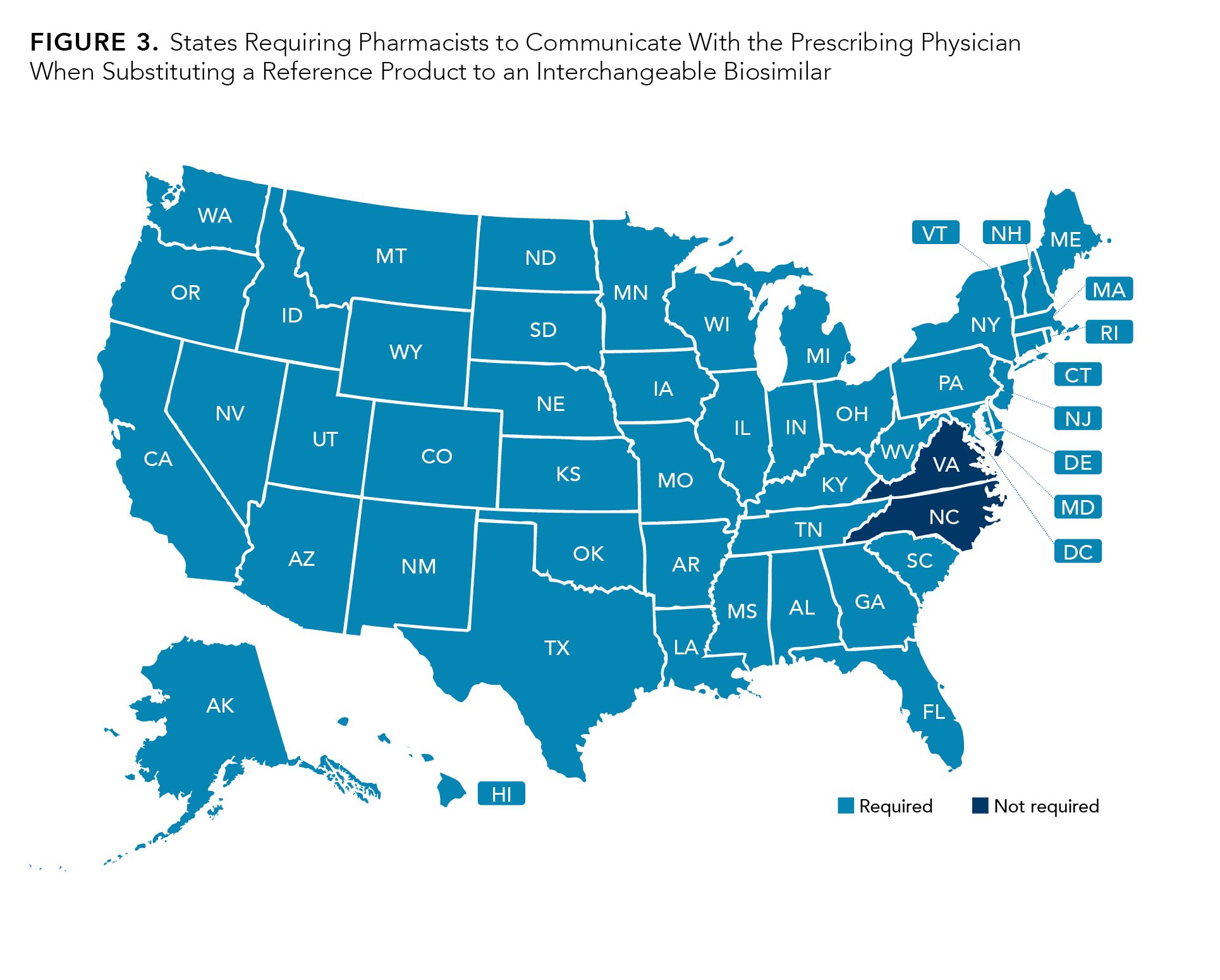
States also differ in terms of the requirements pertaining to communication and record keeping for the pharmacy-level switch from a reference product to an interchangeable biosimilar.21 Nearly all states (48 out of 50) require that the pharmacist communicates back to the prescribing physician when substituting to an interchangeable biosimilar. The states that do not require communication with the prescribing physician are North Carolina and Virginia (Figure 3).


Figure 3.
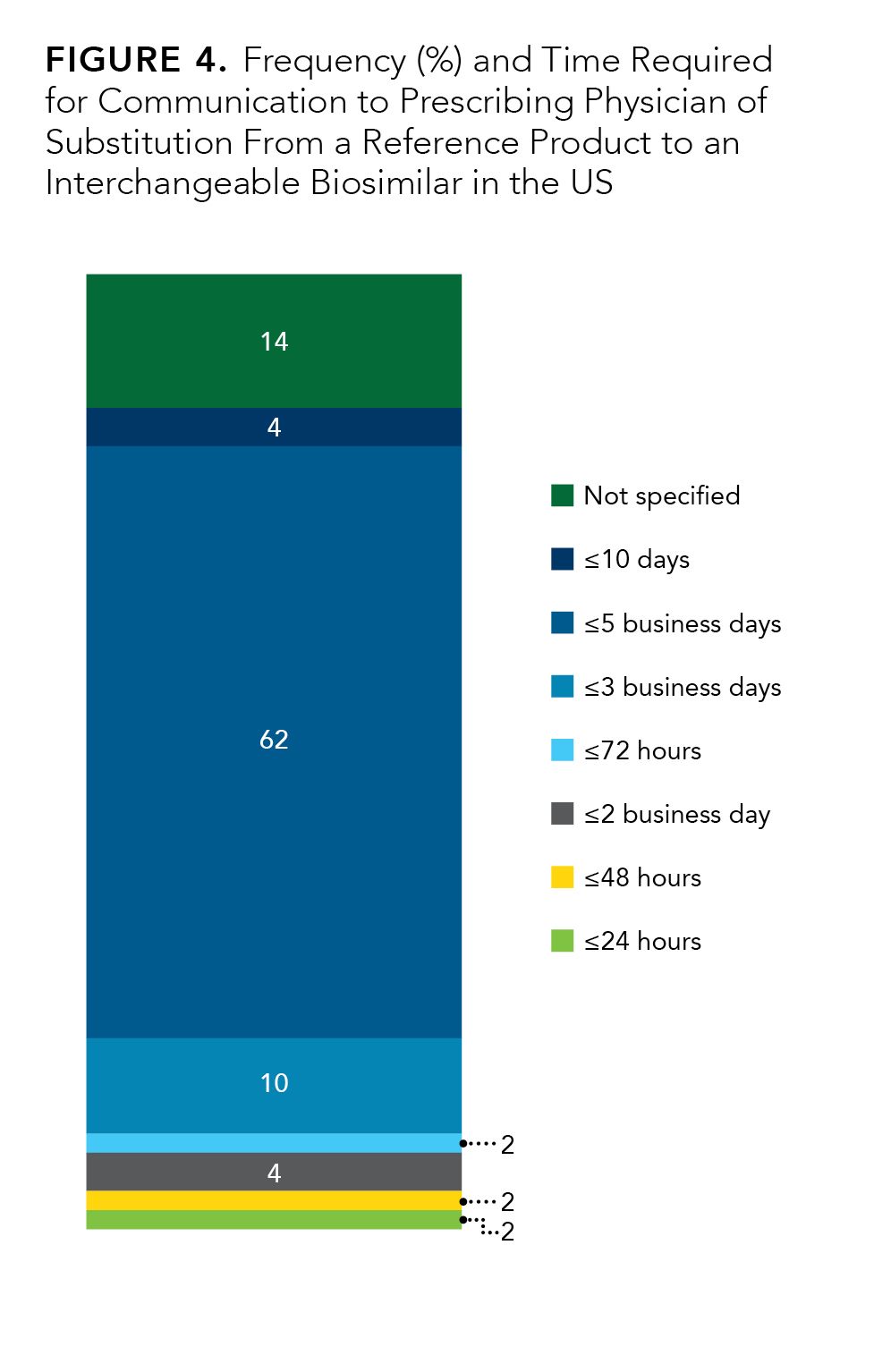
The time frame for such communication varies between states, but the majority (31 out of 50) require that the communication occurs within 5 days of the substitution (Figure 4). Most states have similar requirements regarding communication to patient of the substitution; patient communication is mandatory in 45 states.


Figure 4.
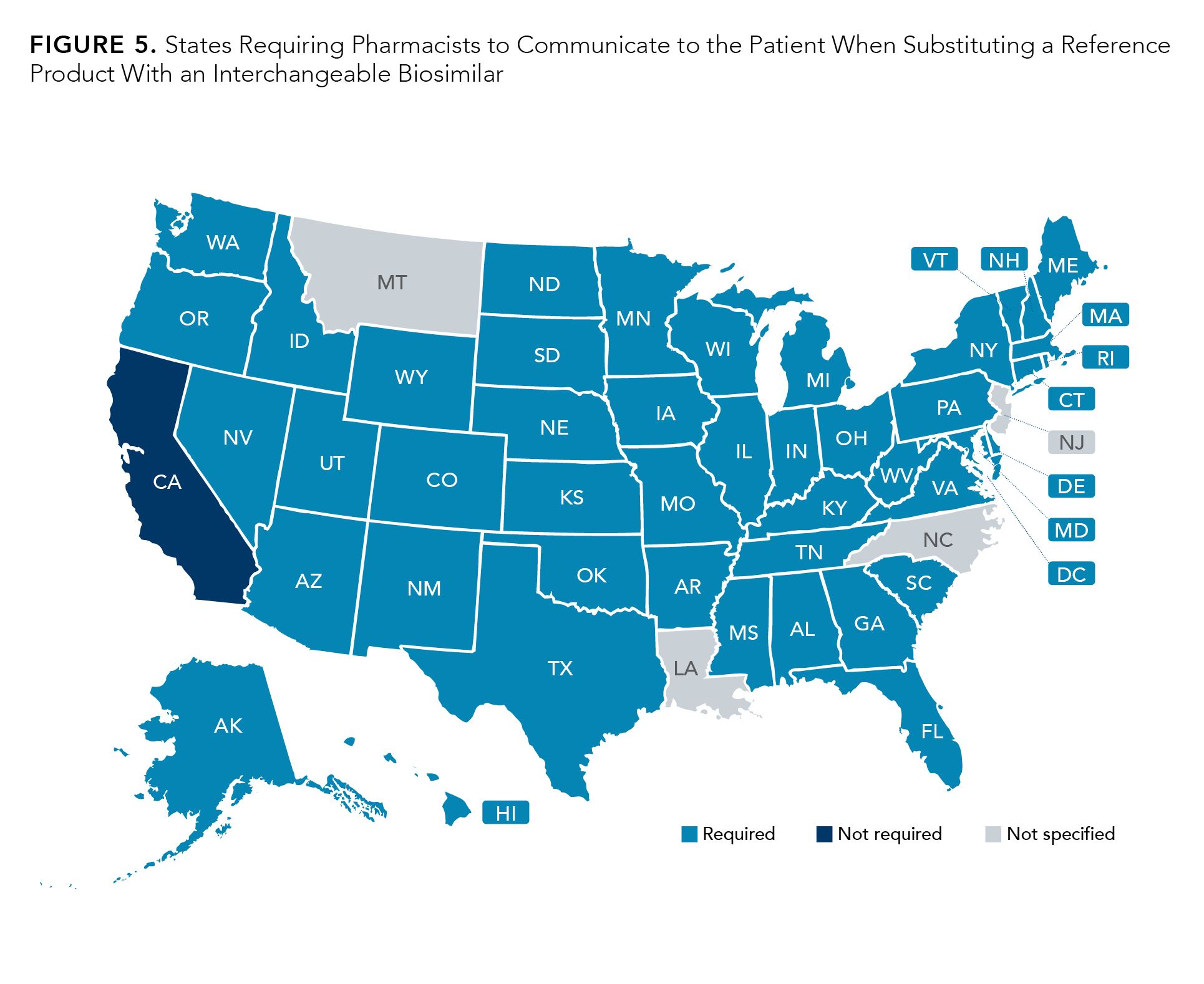
However, Louisiana, Montana, New Jersey, and North Carolina do not specify in the statute, and California does not require such communication at all. (Figure 5).21


Figure 5.
Conclusion
In summary, biosimilars are licensed through an abbreviation approval pathway, and this pathway can involve assessment of interchangeable designation. To be deemed interchangeable, a biosimilar may undergo additional clinical trials to provide evidence that the proposed interchangeable biosimilar will not have diminished efficacy or an increased risk of safety concerns after multiple switches between the reference product and the biosimilar, compared with the reference product alone. The FDA allows pharmacy-level substitution of interchangeable biosimilars, but state-level regulations vary. Most states permit pharmacists to substitute reference biologics with interchangeable biosimilars without authorization from the provider, which is similar to the pharmacy-level substitution of brand-name and generic chemical medications. Because of increased physician confidence and ease of prescription, interchangeable biosimilars may contribute to improved biosimilar uptake in the United States, leading to cost savings for the US health care system.
Author Information. Sophia Humphreys, PharmD, MHA, BCBBS, Sutter Health, Sacramento, California.
Funding Source. This work was supported by Coherus BioSciences, Redwood City, California.
Acknowledgements. Medical writing and editorial assistance were provided by Chantel Kowalchuk, PhD; and Kim Enfield, PhD (ApotheCom, San Francisco, California), with funding provided by Coherus BioSciences (Redwood City, California).
For Correspondence: Sophia Humphreys, PharmD, MHA, BCBBS, Sutter Health, sophia.humphreys@sutterhealth.org
References
1. Jallal B. Realizing the promise of biologics. HUHPR. April 9, 2017. Accessed July 17, 2023. http://www.huhpr.org/volume-16-issue-1-2/2017/4/9/realizing-the-promise-of-biologics
2. Miliotou AN, Papadopoulou LC. CAR T-cell therapy: a new era in cancer immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5-18. doi:10.2174/1389201019666180418095526
3. Bourbeau B, Lyman GH, Lei XJ, et al. Biosimilar use among 38 ASCO PracticeNET practices, 2019-2021. JCO Oncol Pract. 2023;19(7):516-622. doi:10.1200/OP.22.00618
4. Biosimilars in the United States 2023-2027: competition, savings, and sustainability. IQVIA. January 31, 2023. Accessed July 17, 2023. https://www.iqvia.com/insights/the-iqvia-institute/reports/biosimilars-in-the-united-states-2023-2027
5. Biosimilars info sheet. Level 1: foundational concepts. Generics and biosimilars. FDA. Accessed July 17, 2023. https://www.fda.gov/media/154912/download
6. Biosimilars info sheet. Level 1: foundational concepts. Biological product definitions. FDA. Accessed July 17, 2023. https://www.fda.gov/files/drugs/published/Biological-Product-Definitions.pdf
7. Biosimilar basics for patients. FDA. Updated March 21, 2023. Accessed July 17, 2023. https://www.fda.gov/drugs/biosimilars/basics-patients
8. Biosimilars info sheet. Level 1: foundational concepts. Manufacturing and variation. FDA. Accessed July 17, 2023. https://www.fda.gov/media/154913/download
9. Biosimilars info sheet. Level 1: foundational concepts. Biological products, biosimilar products, and interchangeable biosimilar products. FDA. Accessed July 17, 2023. https://www.fda.gov/media/154911/download
10. Sun PD, Foster CE, Boyington JC. Overview of protein structural and functional folds. Curr Protoc Protein Sci. 2004;chapter 17(1):unit 17.1. doi:10.1002/0471140864.ps1701s35
11. What are 351(A) & 351(K)? Freyer Global Regulatory Solutions & Services. 2023. Accessed July 17, 2023. https://www.freyrsolutions.com/what-is-351a-351k
12. Scientific considerations in demonstrating biosimilarity to a reference product. FDA. April 2015. Accessed July 17, 2023. https://www.fda.gov/media/82647/download
13. Hatch-Waxman letters. FDA. Updated February 3, 2022. Accessed July 17, 2023. https://bit.ly/44XGqsZ
14. Tesser JR, Furst DE, Jacobs I. Biosimilars and the extrapolation of indications for inflammatory conditions. Biologics. 2017;11:5-11. doi:10.2147/BTT.S124476
15. Vivian JC. Generic-substitution laws. US Pharm. 2008;33(6):30-34.
16. Considerations in demonstrating interchangeability with a reference product. Guidance for industry. FDA. May 2019. Accessed July 17, 2023. https://www.fda.gov/media/124907/download
17. Eckford C. EMA approves biosimilar interchangeability in EU. European Pharmaceutical Review. September 20, 2022. Accessed July 17, 2023. https://www.europeanpharmaceuticalreview.com/news/174475/ema-approves-biosimilar-interchangeability-in-eu/
18. Barlas S. FDA guidance on biosimilar interchangeability elicits diverse views: current and potential marketers complain about too-high hurdles. PT. 2017;42(8):509-512.
19. Alvarez DF, Wolbink G, Cronenberger C, Orazem J, Kay J. Interchangeability of biosimilars: what level of clinical evidence is needed to support the interchangeability designation in the United States? BioDrugs. 2020;34(6):723-732. doi:10.1007/s40259-020-00446-7
20. US Senate clarifies status of interchangeable biosimilar exclusivity. Generics and Biosimilars Initiative. May 27, 2022. Accessed July 17, 2023. https://bit.ly/3QbXM1m
21. Biosimilar interchangeability laws. Cardinal Health. 2019. Accessed July 17, 2023. https://www.cardinalhealth.com/content/dam/corp/web/documents/publication/Cardinal-Health-Biosimilar-Interchangeability-Laws-by-State.pdf














How Health Care Institutions Can Leverage Biosimilars to Generate Savings
August 17th 2022On this episode of Managed Care Cast, Ryan Haumschild, PharmD, MS, MBA, from Emory Healthcare and the Winship Cancer Institute, explains the evolution of biosimilar pharmacoeconomics and the different strategies that health care institutions can implement to reap the benefits of biosimilar savings.
Listen



