
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Uptake of Oncology Biosimilars: Managed Care Strategies to Improve Value-Based Care Systems
To claim CE credit for this activity, please visit https://www.pharmacytimes.org/courses/uptake-of-oncology-biosimilars-managed-care-strategies-to-improve-value-based-care-systems
Abstract
The use of biosimilars in oncology and as supportive agents for patients with cancer has introduced an important opportunity to expand access to cost-effective care, but their utilization remains inconsistent and is influenced by a variety of factors. Promoting the uptake of biosimilars across healthcare systems relies on improving perception and education about biosimilars, which involves multiple stakeholders, including pharmacists, providers, and patients. Clinicians and managed care professionals must consider comparative analytical studies, clinical efficacy data, and reductions in costs of care associated with biosimilars when establishing protocols for their inclusion within formularies. Real-world switch studies in oncology biosimilars that have demonstrated bioequivalence provide basis to support efficacy and safety to transition to a biosimilar product. Incorporating oncology biosimilars into treatment pathways will be an important next step in providing value-based care to patients with cancer.
Am J Manag Care. 2022;28(suppl 5):S91-S97. https://doi.org/10.37765/ajmc.2022.89188
Introduction
In recent years, the cost of cancer care associated with biologics in the United States has increased at an alarming rate, causing some patients to be priced out of treatment. Biosimilars present important opportunities to lower healthcare costs and expand access to care for oncology patients. Oncology indications and supportive care are leading areas in which the use of biosimilars is rapidly expanding, but it is necessary that clinician and prescriber knowledge increase at the same rate. Multiple factors have caused delays in the incorporation of oncology biosimilars into clinical practice, spanning from legislative restrictions to poor acceptance and misconceptions about biosimilars’ safety and efficacy, which ultimately influence their formulary placement, pricing, and contracting. With improved treatment protocols and therapeutic advances increasing survival rates of patients with cancer and necessitating extended treatment courses, the spotlight has never shone more directly on the importance of incorporating biosimilars into cancer treatment paradigms. Projections estimate that biosimilars will continue to drive price reductions through competition, both with reference agents and among other biosimilars. However, their success depends on consistent use, input from policymakers, and acceptance by patients and prescribers, providing key opportunities for pharmacists to emerge as leaders in biosimilar adoption and education.
Economic Burden of Cancer and Biologics in the United States
Current projections for numbers of new cancer diagnoses and deaths in the United States do not yet account for the impact of the COVID-19 pandemic but estimate more than 1.9 million new cancer cases and approximately 610,000 cancer deaths to occur in 2022.1 Biologic drugs have become fundamental in the treatment of cancer, and their continued growth is expected over the next decade due to increased rates of cancer screenings, more effective treatments, and survivorship.2,3 However, the regulatory pathways for biologics are complex and require lengthy, in-depth preclinical and clinical trials, which increase their cost.4,5 Based on 2019 sales data, monoclonal antibody-based biologics, including rituximab, bevacizumab, and trastuzumab, play a major role in the treatment of patients with cancer. The use of granulocyte colony-stimulating factors (GCSFs) filgrastim and pegfilgrastim also increased for patients receiving high-risk febrile neutropenia (FN) chemotherapy regimens. From 2014 to 2019, the use of GCSFs increased from 75% to 83% and 75% to 86% in commercially insured and Medicare patients, respectively.6 The costs associated with the development process of biologic drugs are dependent on manufacturing conditions, with prices increasing after market entry by up to 25% on average.7,8 As the most frequently used agents in cancer care, biologics represent just 2% of total prescriptions in the United States yet account for 37% of total drug expenditures.9The total global spend on cancer treatments was calculated at $150 billion in 2018, with a projected increase to $240 billion by 2023.8
The opportunity to combat the high cost of oncology biologics exists in the production of biosimilars, or agents comparable in clinical efficacy. Biosimilars are developed from natural (biologic) sources and are licensed under an abbreviated 351(k) licensure pathway established by The Biologics Price Competition and Innovation Act (BPCIA) of 2009. The BPCIA permits manufacturers to seek approval of a biosimilar using already proven clinical efficacy and safety data of its reference product, which eliminates duplication of clinical trials, saving time and resources compared with development of the biologic reference product.10 Because of the complexity of these molecules, biosimilars are not identical to their reference product, and there may be slight differences in regard to structure. Prior to U.S. Food and Drug Administration (FDA) approval, biosimilars must prove no clinically meaningful differences in purity or bioactivity when compared with the reference biologic.11
A “totality of evidence” approach is required for each biosimilar under development and is determined in a stepwise manner through extensive comparative analytical studies with a reference product. The primary objective of these comparative trials is to confirm mechanism of action, analyze structure and function, and the safety profile of the biosimilar when compared with the reference product. This differs from the approval process for generic medications, as the only requirement is that the generic product demonstrates bioequivalence with the brand-name product. As there may be slight structural differences, biosimilars are extensively evaluated for immunogenicity, or the potential for impurities or small changes occurring during the manufacturing process to elicit a severe adverse reaction or immunogenic response in patients that was not observed with use of the reference product.11 To date, there have been no immunogenicity concerns for any FDA-approved oncology biosimilars.12
With the expected expiration of several patents on oncology biologics, including pertuzumab and ipilimumab, between 2023 and 2027, there is opportunity for an increase in the launch of biosimilars to market in the coming years with vast economic implications, and the BPCIA is essential in expediting market entry of biosimilars.13,14 However, 2 important factors influencing future uptake of biosimilars are interchangeability and extrapolation. Manufacturers of biosimilars seeking an interchangeable designation or to extrapolate use in other indications approved for the reference product must meet additional safety and efficacy standards in clinical trials, demonstrate the same clinical results as the reference product, and show no greater risk or decreased efficacy when switching between the products in any patient. An interchangeable designation would allow for the automatic substitution of the biosimilar for the reference product without prescriber intervention. Currently, just 2 biosimilars have been approved as interchangeable, though neither have oncology indications. Future biosimilars that meet interchangeable requirements will be key to increasing uptake, as 70.6% of physicians reported interchangeability would favorably influence their views of biosimilars.15 Until an interchangeable oncology biosimilar is approved, pharmacy and therapeutics committees will be responsible for evaluating the totality of evidence for biosimilars and can deem them to be therapeutically equivalent with their reference products for some or all approved indications.16
The ability to switch drug therapy in patients with agents that are therapeutically equivalent is an important capability for pharmacists and is central to reducing costs and increasing drug access for patients. The FDA allows for biosimilars to be approved for indications of the reference product without direct studies in that indication through a concept of extrapolation, which is based on currently available data for the biosimilar as well as previous efficacy and safety data for the reference product.11 Extrapolating would offer the potential for biosimilar products to be used for additional tumor types for which the reference product is licensed, thus improving medication access for patients without the need to replicate indication-specific clinical studies with already established data.11,16,17 However, the lack of clinical trial data has led to initial pushbacks from the provider community. When biosimilars first launched, immunogenicity was a major concern, especially in the autoimmune space. Extrapolation increased uncertainties around potential immunogenicity in non-studied indications. Recent data have lightened up such concerns showing no significant immunogenicity differences between biosimilars and reference products.18 As familiarities with biosimilar products increased, extrapolation of indications has been increasingly accepted in clinical practice, leading to more uptake of biosimilar products overall. Despite increased acceptance, extrapolation of indications was reported by 30% of payers to be a key factor influencing inclusion of biosimilars in formulary.15 This is intrinsically different from a designation of interchangeable, as substitution with therapeutically equivalent medications may require prescriber notification according to institution protocols. However, biosimilar substitution has specific requirements and laws that vary state-by-state that pharmacists and prescribers must keep in mind.16
The Economic Benefits of Biosimilars in Reducing Overall Cost of Cancer Treatment
The 3 biosimilars for oncology biologics launched in 2019 achieved significant uptake within their first year, with bevacizumab leading at 42%, trastuzumab at 38%, and rituximab at 20%, and their use continued to increase at rates higher than observed with earlier biosimilars.19 Similar uptake was observed with the initial filgrastim biosimilar, filgrastim-sndz, approved in 2015 and pegfilgrastim biosimilars pegfilgrastim-jmdb and pegfilgrastim-cbqv approved in 2018. Uptake of filgrastim-sndz was rapid and accounted for 47% and 42% of filgrastim use among commercially insured and Medicare populations in 2018, respectively; previously mentioned pegfilgrastim biosimilars combined to account for 29.8% of all long-acting GCSF use among commercially insured patients in 2019.6,20 The oncology biosimilars with indications for treatment or supportive care currently FDA approved as of June 1, 2022, are listed in the Table.21,22


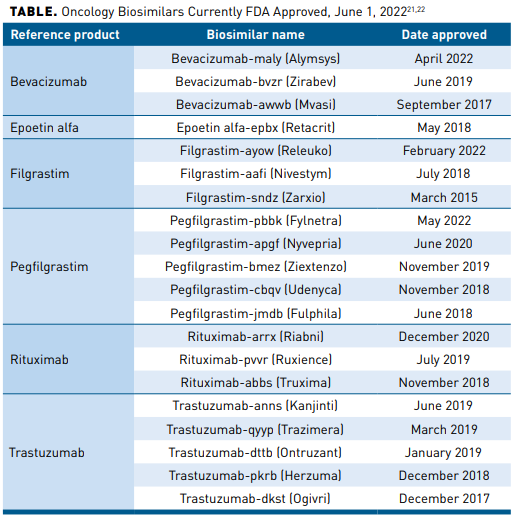
The continued development, approval, and adoption of oncology biosimilars is essential to lower costs, expand patient access to care, and increase economic pressure on manufacturers. Biosimilars are priced, on average, between 15% and 35% lower than their reference products, with an estimated projected overall savings of $100 billion over the next 5 years and $13.5 billion over the next 10 years.12,19,23,24 The economic benefit of biosimilars may also be passed directly on to patients in the form of reduced out-of-pocket costs, with patients who received a biosimilar paying on average 12% to 45% less out of pocket than those receiving a reference product.25 Based on a study conducted by the Bloomberg School of Public Health at Johns Hopkins University that surveyed large employer groups, patients who took the biosimilar paid on average 45%, approximately $600, less for filgrastim out of pocket than those who took the originator product.25 In the case of bevacizumab, one study determined a total cost savings of $3,430,967 in year 1 and $14,731,112 in year 3 in a hypothetical 10 million-member health plan when patients were switched from the reference product to the bevacizumab biosimilar; more than half of costs were attributed to patients with colorectal cancer.8 Mayo Clinic recently published their savings results from initiating a biosimilar first strategy. Utilization increase of preferred biosimilars was 69% for bevacizumab, 63% for epoetin alfa, 80% for filgrastim, 79% for rituximab, and 72% for trastuzumab. Savings assessed at 12 months post implementation were $23.1 million.26
Switching Studies in Oncology Biosimilars: Real-World Evidence
Despite recent advancements and approvals of biosimilars, data on their use in the oncology setting in the United States remain scarce. Available analyses, such as from Flatiron Health’s electronic health record (EHR) database, indicate the overall utilization rate of rituximab-pvvr, trastuzumab-anns, and bevacizumab-awwb is increasing among patients with cancers.27-29 Real-world adoption has aligned with current guideline recommendations, with one study reporting rituximab-pvvr was frequently administered as part of a combination regimen for patients who were switched.27
Both the trastuzumab-anns and bevacizumab-awwb biosimilars demonstrated bioequivalence to their reference products, allowing for extrapolation and approval for additional indications of their reference products. According to the Flatiron Health EHR database, in the first 12 months post launch, the bevacizumab-awwb biosimilar was integrated into patient treatment plans within 2 weeks of market availability, and approximately 50% of patients receiving reference bevacizumab were transitioned to the biosimilar within 28 days, with 83% of treatment-experienced patients switched without disease progression.28,29 Bevacizumab-awwb was initiated as first-line therapy within a median of 63 days in 91% of patients who were naïve to reference bevacizumab.28
Similarly, trastuzumab-anns was integrated within 3 weeks (earliest use at 4 days post market entry), and the majority of patients receiving the trastuzumab reference product were transitioned to the biosimilar within 28 days, possibly indicative of overall familiarity with protocols and increased confidence among physicians and prescribers with use of these biosimilars. A significant proportion of patients who were previously treated with reference products were under financial burden and receiving support, which could have contributed to providers’ decisions to initiate biosimilars to relieve patients of financial toxicities.29
Cost-Effectiveness of Oncology Biosimilars in Supportive Care
In addition to oncology biosimilars indicated for the treatment of cancer, use of biosimilars for supportive cancer care, such as hematopoietic growth factor or GCSF, have also been associated with significant cost savings and increased access to treatment.30 The availability of biosimilars for supportive oncology agents has increased access to treatment and reduced costs for patients. In general, switching from use of pegfilgrastim to its biosimilar has been associated with a 30% or greater cost reduction.31 In 2 separate analyses, switching to pegfilgrastim-cbqv has been shown to expand patient access to much-needed R-CHOP therapy for patients with non-Hodgkin lymphoma (NHL) and to FOLFIRINOX for patients with metastatic pancreatic cancer.30,32 Switching a population of 20,000 patients from reference pegfilgrastim to pegfilgrastim-cbqv resulted in per-patient savings of $806 per cycle for a total of $4833 for 6 cycles of therapy.30 Another analysis investigated savings to drug acquisition costs for pegfilgrastim-cbqv compared with its reference product in patients with metastatic pancreatic cancer and determined per-patient savings of $575.62 for 1 cycle and $6907.41 for 12 cycles.32 These findings are significant for patients receiving long-term maintenance therapies, and savings may also offset costs associated with newer oncology treatments, such as chimeric antigen receptor T-cell therapy or targeted therapies.4 The reported savings associated with utilization of pegfilgrastim-cbqv in one patient with metastatic pancreatic cancer would allow for 2 additional cycles of FOFIRINOX for one patient; if 9 patients with NHL were converted to pegfilgrastim-cbqv for 6 cycles, one patient would receive 6 cycles of R-CHOP therapy.30,32
Challenges Affecting Uptake of Oncology Biosimilars in Clinical Practice
Though the concept of biosimilars is becoming more familiar to patients and providers, there are differences regarding the approval and authorization of biosimilars between regulators in Europe and the United States, which can cause confusion and may partially explain why biosimilar uptake and usage in clinical practice in the United States continues to lag.33 Biosimilars have experienced rapid uptake in European countries due to financial incentives for increased use.3 But, in the United States, biosimilar utilization is inconsistent and is influenced by differences in formularies as well as prescriber and physician experience, judgment, and level of comfort. Overwhelmingly, data indicate that prescribers are aware of the economic benefits of biosimilars, with financial savings to the patient often reported as the most influential factor in their willingness to prescribe a biosimilar.34 However, patients and physicians have discordant perspectives of biosimilar safety and efficacy, perceiving biosimilar agents to be cheaper, less effective, or associated with more adverse effects.34 When presented with prescribing and treatment scenarios, 86% of respondents indicated they would choose the reference product over the biosimilar when both were available, regardless of whether patients had experience with or were naïve to the drug product; when only the biosimilar was available on formulary, 83% of prescribers recommended the biosimilar when patients were naïve to the products and 80% recommended the biosimilar if the patient had prior experience with the reference product.34 Though the approval of biosimilars for additional indications via extrapolation has increased access for patients, the lack of specific clinical data in these indications cannot be overlooked and has limited use of biosimilars in extrapolated indications.35
Though physicians consistently report high levels of confidence in the safety and efficacy of biosimilars compared with their reference products, these beliefs do not correspond to observed prescribing trends in clinical practice.15 One survey found trastuzumab, rituximab, and bevacizumab reference products to be in the non-preferred position in 70% of managed care organizations’ (MCOs) formularies, though all provided some coverage for biosimilars of reference products.15 In these scenarios, physicians have reported switching from reference product to biosimilar in less than 25% of patients who have been receiving treatment with the reference product.15 In another survey of 86 International Society of Oncology Pharmacy Practitioners (ISOPP) members, participants identified top areas for learning needs, including evaluating efficacy of the biosimilar compared with the reference product (74.4%), managing switching from reference product to biosimilar (74.4%), and limited knowledge of safety of biosimilars (73.3%).36 Moreover, 64% of ISOPP respondents stated their decision to use biosimilar products was dependent upon institutional policies, which opens the possibility for pharmacists involved in formulary decision making to increase inclusion of biosimilars.36
The last population that can influence uptake of biosimilars is patients with cancer. Patients have a right to be informed about their treatment, including any viable alternative therapies, such as biosimilars.37 If patients do not fully understand or receive misconstrued information from clinicians about safety and efficacy of biosimilars, they may be more likely to request the reference product. Reliable sources of biosimilar information have become readily available with the increase in approvals and market availability, but messaging will remain inconsistent if they are not utilized by healthcare providers. This provides a key opportunity for pharmacist involvement in providing physician and patient education to aid in the understanding and successful uptake of oncology biosimilars.
Making Oncology Biosimilars More Accessible: Inclusion in Formularies, Pathways, and Protocols
There remains significant untapped potential for the use of biosimilars in the oncology setting. As additional biosimilars gain approval, pharmacy and therapeutics committees will become increasingly important in ensuring patient access and must weigh the totality of clinical evidence and safety data when evaluating their inclusion within formularies.16 In addition, decision makers must consider the primary patient population(s) within the institution. Committees may also need to consider extrapolation data in their formal review when deciding which indications to approve or restrict use of a biosimilar. As more data for biosimilars become available, postmarketing studies with real-world evidence will become increasingly important in establishing safety and efficacy of biosimilars. Finally, the cost of the medication combined with ancillary services related to the biosimilar must be analyzed, such as maintaining adequate pharmacy stock of all biosimilars for a reference product if one is not preferred.16
At the hospital system level, medication reimbursement and billing policies remain areas in which reforms are needed to increase adoption of oncology biosimilars in clinical practice.27 Discrepancy in biosimilar uptake between physician offices and hospital outpatient departments could be the result of several factors, including different net prices resulting from manufacturers’ price concessions, complex decision making among hospital systems, and coverage restrictions that may stem from the institution’s primary patient population.38 Additionally, markups for reference biologics are reported to be between 116% and 121% in hospital outpatient department settings compared with physician offices (18%-19%). Biosimilars bevacizumab-awwb and trastuzumab-anns were marked up at 32% and 35%, respectively, in physician offices.3
Until an interchangeable oncology biosimilar is approved, payers may continue to make product-by-product decisions before adding a biosimilar to system formularies. “Step therapy” requirements are often implemented, meaning that a preferred agent must be tried before a non-preferred agent to ensure insurance coverage.39 In the oncology space, it is more widely acceptable to introduce biosimilars into the treatment plans for treatment-naïve patients, with physicians and practice managers reporting greater than 50% utilization of biosimilars among patients who were naïve to reference trastuzumab, rituximab, and bevacizumab.15 A recent analysis of commercial, Medicare, and Medicaid health plan claims data reported 44% of payers implemented a biosimilar step therapy protocol for oncology patients initiating therapy and 15% have initiated for both treatment-naïve and treatment-experienced patients.40 It may be more difficult to use these agents for those who are mid-treatment or are stable in their current treatment protocol. Payers additionally reported cost differential associated with use of a biosimilar agent (average 27%) to be a leading factor (88%) in establishing criteria for biosimilar step therapy protocols.40
Payer Strategies to Encourage Uptake of Oncology Biosimilars
Given the lower costs of biosimilars, they are likely to play a significant role in value-based care systems, and it is expected that increased availability of biosimilars coming to market in the coming years will continue to spur price competition. Government, integrated delivery networks, and accountable care organizations (ACOs) may be involved in pricing and formulary negotiations, where a discount may be given for formulary placement. ACOs have significant bargaining power and will likely influence decisions among prescribers and institutions to use biosimilars in place of the reference biologic, which can lead to price competition at institution level for pricing set by manufacturers. In many settings, because there are no clinically significant differences between a biosimilar and its reference product, primary patient populations, manufacturer incentives, and payer reimbursement policies often guide decision making, but discussions should include insight from multiple stakeholders, including pharmacists.9,38 In a survey of payers, two-thirds indicated they had established preferred oncology biosimilars versus the reference products.41 However, payer preference can often change, and differences between formularies can further complicate biosimilar utilization and uptake.42 Furthermore, additional data are needed to show switching between biosimilars is acceptable clinically.
Medicare Part D does not include biosimilars under the Medicare Coverage Gap Discount Program, making these patients ineligible for the 50% discount on brand-name drugs and brand-name biologics.12 This practice adversely affects seniors and persons with disabilities potentially having higher out-of-pocket costs, especially considering the population of patients older than 60 years is at highest risk for developing invasive cancer.1 Important considerations for payers will be creation and implementation of policies that increase patient access to biosimilars and decrease overall cost of care. Lower costs are additionally likely to improve patient compliance with therapy, thus simultaneously improving outcomes of care and the value of a biosimilar to MCOs.
Future Directions Promoting Access to Care With Biosimilars
In February 2020, the FDA and Federal Trade Commission issued a joint statement to improve access to information about biosimilars, limit spread of misinformation about biosimilars, and advocate for greater competition and availability of biosimilars as they come to market to reduce financial burden for patients.43 Improving patient and prescriber confidence in the use of biosimilars will be critical in their adoption, and several guidelines and statements have been issued to reinforce essential factors in the development of biosimilars, such as the importance of clinical evidence and regulatory review periods to establish biosimilar safety and efficacy.8,44 Additionally, in April 2021, the US Congress passed the Advancing Education on Biosimilars Act of 2021, allowing the FDA along with the Department of Health and Human Services to formally advance and provide education for healthcare providers, patients, and caregivers regarding biosimilar products and abilities of pharmacists to engage in biosimilar substitution.24,45 A recent report from the American Society of Clinical Oncology clarifies definitions and previous statements on the place of oncology biosimilars in therapy as well as offers updates to recommendations published prior to market entries of rituximab-pvvr, trastuzumab-anns, and bevacizumab-awwb in 2019.12 Positioned on the oncology care team and within formulary committees, managed care pharmacists will be essential in the development and end use of these resources.
Biosimilars have the potential to counter rising costs in oncology care. To increase preference for utilization of lower cost medications and achieve goals of value-based provision of care, alternative reimbursement models that shift economic incentives are necessary. Current “buy-and-bill” method is not an incentive for providers to utilize biosimilars, as these reimbursement models reimburse neutrally based on average sales price (ASP) of the reference product. Payers could alter the fee schedule and increase reimbursement for the biosimilars to create a natural gravitation towards the preferred biosimilar products.
Unlike healthcare models in Europe where national policies have been adopted to encourage and incentivize biosimilar usage, the United States does not have a national healthcare system. Pharmacy benefit managers and third-party payers may impose pressures on manufacturers to reduce costs further through rebates and discounts. Medicare Part B reimbursement value for a biosimilar is based on the drug’s ASP plus 6% of the reference product, although other reimbursement strategies have been reported based on average wholesale price or at the maximum allowable cost.9,40 Additional considerations for reimbursement in models where value is based on ASP of each individual biosimilar plus 6% ASP of reference product or in reimbursement based on combined ASP of reference and biosimilar products may present a strong economic incentive for the improved utilization of biosimilars.3,12 For private insurance, it is dependent on preferred products. An alternative reimbursement model proposed by Yang, et al, accounted for the greatest savings in year 1 after market entrance of biosimilars for trastuzumab, bevacizumab, and rituximab in both physician office and hospital outpatient department settings; the decline in savings projected across years 2 through 5 was attributed to potential of increasing biosimilar market share.3
Biosimilars will have a growing role within evolving frameworks to offer value-based care to patients receiving supportive care or treatment for cancer. Within value-based care programs developed by the Centers for Medicare & Medicaid Services, oncology care models incorporate standard monthly enhanced oncology services payments with prespecified performance-based metrics that tie to provision of higher quality, more efficient, and lower cost patient care.46 As participants in these qualitative measures, pharmacist input can be integral in identifying opportunities to improve delivery of care while controlling costs for patients, and the cost savings resulting from utilization of biosimilars can assist in achieving these goals.
Conclusions
There are vast opportunities for the continued utilization of oncology biosimilars, but consistent use and considerations regarding interchangeability and switching have affected optimal incorporation of biosimilars into formularies and institution protocols. Ultimately, evaluation of overall costs and cost savings will be key to the consistent adoption of biosimilars in clinical practice. Negotiations between payers and manufacturers will likely continue as competition for formulary approval increases, but failure to incorporate biosimilars into cancer treatment plans is a disservice to patients and the healthcare system. It is inevitable that the role of pharmacists in advising, providing education, and ensuring the safe use of biosimilars will grow alongside future approvals of biosimilars with oncology indications. Educating other healthcare providers to select treatments that will provide the greatest value to patients is a significant step in facilitating the clinical uptake and usage of biosimilars.
Author affiliation: YuQian Liu, PharmD, is Director, Specialty Clinical Solutions, Magellan Rx Management, Middletown, RI.
Funding source: This activity is supported by an educational grant from Coherus BioSciences.
Author disclosure: Dr Liu has no relevant financial relationships with commercial interests to disclose.
Authorship information: Concept and design, analysis and interpretation of data, critical revision of the manuscript for important intellectual content.
Address correspondence to: yuliu@magellanhealth.com
Medical writing and editorial support provided by: Brianna Winters, MA
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7-33. doi: 10.322/caac.21708
- Bottom D, Davies C. Biosimilars to continue rapid growth over the next decade. IQVIA Blog. Published January 3, 2022. Accessed May 19, 2022. www.iqvia.com/blogs/2021/12/biosimilars-to-continue-rapid-growth-over-the-next-decade
- Yang J, Carioto J, Pyenson B, et al. Greater uptake, an alternative reimbursement methodology needed to realize cost-saving potential of oncology biosimilars in the United States. J Manag Care Spec Pharm. 2021;27(12):1642-1651. doi: 10.18553/jmcp.2021.21202
- Hubel K, Kron F, Lux MP. Biosimilars in oncology: effects on economy and therapeutic innovations. Eur J Cancer. 2020;139:10-19. doi: 10.1016/j.ejca.2020.07.037
- Blevins Primeau AS. Biosimilar therapies for cancer. Cancer Therapy Advisor. Published July 26, 2019. Accessed May 19, 2022. cancertherapyadvisor.com/home/tools/fact-sheets/biosimilar-therapies-for-cancer-treatment-patient-fact-sheet/2/
- Wang CY, Heldermon CD, Vouri SM, et al. Trends in use of granulocyte colony-stimulating factor following introduction of biosimilars among adults with cancer and commercial or Medicare insurance from 2014 to 2019. JAMA Netw Open. 2021;4(11):e2133474. doi: 10.1001/jamanetworkopen.2021.33474
- Gordon N, Stemmer SM, Greenberg D, Goldstein DA. Trajectories of injectable cancer drug costs after launch in the United States. J Clin Oncol. 2018;36(4):319-325. doi: 10.1200/JCO.2016.72.2124
- Peeters M, Planchard D, Pegram M, Goncalves J, Bocquet F, Jang H. Biosimilars in an era of rising oncology treatment options. Future Oncol. 2021;17(29):3881-3892. doi: 10.2217/fon-2021-0546
- Ngo D, Chen J. A clinical review of biosimilars approved in oncology. Ann Pharmacother. 2021;55(3):362-377. doi: 10.1177/1060028020944596
- Implementation of the Biologics Price Competition and Innovation Act of 2009. U.S. Food and Drug Administration. Updated February 12, 2016. Accessed May 19, 2022. www.fda.gov/drugs/guidance-compliance-regulatory-information/implementation-biologics-price-competition-and-innovation-act-2009
- Biosimilar development, review, and approval. U.S. Food and Drug Administration. Updated October 20, 2017. Accessed May 19, 2022. www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval
- Nahleh Z, Lyman GH, Schilsky RL, et al. Use of biosimilar medications in oncology. JCO Oncol Pract. 2022;18(3):177-186. doi: 10.1200/OP.21.00771
- Ditani AS, Mallick PP, Anup N, et al. Biosimilars accessible in the market for the treatment of cancer. J Control Release. 2021;336:112-129. doi: 10.1016/j.jconrel.2021.06.014
- Goyal D, Nidhi. List of biologic coming off patents between 2022 to 2027. GreyB Services. Accessed May 19, 2022. www.greyb.com/biologics-patents-expiring-2022-2023-2024-2025-2026-2027/
- Yang J, Blinzler K, Lankin J, Vijayakumar S, Maculaitis MC, Shelbaya A. Evolving perceptions, utilization, and real-world implementation experiences of oncology monoclonal antibody biosimilars in the USA: perspectives from both payers and physicians. BioDrugs. 2022;36(1):71-83. doi: 10.1007/s40259-021-00509-3
- Cuellar S, McBride A, Medina P. Pharmacist perspectives and considerations for implementation of therapeutic oncology biosimilars in practice. Am J Health Syst Pharm. 2019;76(21):1725-1738. doi: 10.1093/ajhp/zxz190
- Biosimilar and interchangeable biologics: more treatment choices. U.S. Food and Drug Administration. Updated October 12, 2021. Accessed May 19, 2022. www.fda.gov/consumers/consumer-updates/biosimilar-and-interchangeable-biologics-more-treatment-choices
- Strand V, Goncalves J, Hickling TP, Jones HE, Marshall L, Isaacs JD. Immunogenicity of biosimilars for rheumatic diseases, plaque psoriasis, and inflammatory bowel disease: a review from clinical trials and regulatory documents. BioDrugs. 2020;34(1):27-37. doi: 10.1007/s40259-019-00394-x
- Biosimilars in the United States 2020-2024: competition, savings, and sustainability. IQVIA. Published September 29, 2020. Accessed May 19, 2022. www.iqvia.com/insights/the-iqvia-institute/reports/biosimilars-in-the-united-states-2020-2024
- Karaca-Mandic P, Chang J, Go R, Schondelmeyer S, Weisdorf D, Jeffrey MM. Biosimilar filgrastim uptake and costs among commercially insured, Medicare Advantage. Health Aff (Millwood). 2019;38(11):1887-1892. doi: 10.1377/hlthaff.2019.00253
- Biosimilar product information. U.S. Food and Drug Administration. Updated February 25, 2022. Accessed May 19, 2022. www.fda.gov/drugs/biosimilars/biosimilar-product-information
- Brennan Z. US biosimilar launches about to turn a corner. Regulatory Affairs Professionals Society. March 16, 2020. Accessed May 19, 2022. www.raps.org/news-and-articles/news-articles/2019/12/us-biosimilar-launches-about-to-turn-a-corner
- San-Juan-Rodriguez A, Gellad WF, Good CB, Hernandez I. Trends in list prices, net prices, and discounts for originator biologics facing biosimilar competition. JAMA Netw Open. 2019;2(12):e1917379. doi: 10.1001/jamanetworkopen.2019.17379
- Becker C. Decreasing drug costs through generics and biosimilars. National Conference of State Legislatures. Published January 21, 2022. Accessed May 19, 2022. www.ncsl.org/research/health/decreasing-drug-costs-through-biosimilars.aspx
- Socal M, Ballreich J, Chyr, L, Anderson G. Biosimilar medications – savings opportunities for large employers. Department of Health Policy and Management, Johns Hopkins Bloomberg School of Public Health. Published March 2020. Accessed May 19, 2022. www.eric.org/wp-content/uploads/2020/03/JHU-Savings-Opportunities-for-Large-Employers.pdf
- Jensen CJ, Ticky EM, Lempke MB, et al. Implementing and optimizing biosimilar use at Mayo Clinic. Mayo Clin Proc. 2022;S0025-6196(22)00119-7. doi: 10.1016/j.mayocp.2022.02.015
- Shelbaya A, Kelton JM, Thompson J, Alvir JMJ, Maculaitis MC, Yang J. Real-world use and acceptance of biosimilar monoclonal antibodies of rituximab in oncology practice in the USA. Future Oncol. 2021;17(30):3941-3950. doi: 10.2217/fon-2021.0618
- Rhodes W, DeClue RW, Accortt N, et al. Real-world use of bevacizumab-awwb, a bevacizumab biosimilar, in US patients with metastatic colorectal cancer. Future Oncol. 2021;17(36):5119-5127. doi: 10.2217/fon-2021-0588
- Jin R, Mahtani RL, Accortt N, Lawrence T, Sandschafer D, Loaiza-Bonilla A. Clinical and treatment characteristics of patients treated with the first therapeutic oncology biosimilars bevacizumab-awwb and trastuzumab-anns in the US. Ther Adv Med Onc. 2021;13:17588359211041961. doi: 10.1177/17588359211041961
- McBride A, MacDonald K, Abraham I. Conversion to supportive care with biosimilar pegfilgrastim-cbqv enables budget-neutral expanded access to R-CHOP treatment in non-Hodgkin lymphoma. Leuk Res. 2021;106:106591. doi: 10.1016/j.leukres.2021.106591
- Cornes P, Gascon P, Vulto AG, Aapro M. Biosimilar pegfilgrastim: improving access and optimizing practice to supportive care that enables cure. BioDrugs. 2020;34(3):255-263. doi: 10.1007/s40259-020-00411-4
- MacDonald K, Alrawashdh N, McBride A, Abraham I. Conversion to biosimilar pegfilgrastim-cbqv enables budget-neutral access to FOLFIRINOX treatment for metastatic pancreatic cancer. Future Oncol. 2021;17(33):4561-4570. doi: 10.2217/fon-2021-0718
- Soares JCS, Cavalcanti IDL, de Albuquerque Vasconcelos JL. Can biosimilar products be interchangeable? Pharmaceutical perspective in the implementation of biosimilars in oncology. J Oncol Pharm Pract. 2021;27(6):1491-1502. doi: 10.1177/10781552211016099
- Kolbe AR, Kearsley A, Merchant L, et al. Physician understanding and willingness to prescribe biosimilars: findings from a US national survey. BioDrugs. 2021;35(3):363-372. doi: 10.1007/s40259-021-00479-6
- Gascon P, Krendyukov A, Mathieson N, Natek M, Aapro M. Extrapolation in practice: lessons from 10 years with biosimilar filgrastim. BioDrugs. 2019;33(6):635-645. doi: 10.1007/s40259-019-00373-2
- Chan A, Patel H, Siderov J, Bubalo J, Foreman E. Assessing biosimilar education needs among oncology pharmacy practitioners worldwide: an ISOPP membership survey. J Oncol Pharm Pract. 2020;26(3_suppl):11-21. doi: 10.1177/1078155219898510
- Michels D, Cahill M. Informed consent and chemotherapy. J Oncol Pract. 2005;1(3):99. doi: 10.1200/jop.2005.1.3.99
- Socal MP, Anderson KE, Sen A, Bai G, Anderson GF. Biosimilar uptake in Medicare Part B varied across hospital outpatient departments and physician practices: the case of filgrastim. Value Health. 2020;23(4):481-486. doi: 10.1016/j.jval.2019.12.007
- Use of step through policies for competitive biologics among commercial US insurers. Avalere Health. Published May 7, 2018. Accessed May 19, 2022. avalere.com/insights/use-of-step-through-policies-for-competitive-biologics-among-commercial-us-insurers
- Medical pharmacy trend report. Magellan Rx Management; 2020. Accessed May 19, 2022. www1.magellanrx.com/documents/2021/05/2020-mrx-medical-pharmacy-trend-report.pdf/
- Clark J, Fortier K, Knoblauch B, Reddan J, Jackson T, Kidder P. Payer perceptions and practices regarding oncology therapy management. Blue Fin Group. Posted October 27, 2021. Accessed May 19, 2022. https://consultbfg.com/payer-perceptions-and-practices-regarding-oncology-therapy-management/
- Oskouei ST. Biosimilars: what we can learn from early adopters. Cardinal Health. Accessed May 19, 2022. www.cardinalhealth.com/content/dam/corp/web/documents/whitepaper/cardinal-health-biosimilars-adoption-in-oncology.pdf
- FTC, FDA sign joint statement promoting competition in markets for biologics. Press release. Federal Trade Commission; February 3, 2020. Accessed May 19, 2022. ftc.gov/news-events/news/press-releases/2020/02/ftc-fda-sign-joint-statement-promoting-competition-markets-biologics
- Lyman GH, Balaban E, Diaz M, et al. American Society of Clinical Oncology Statement: biosimilars in oncology. J Clin Oncol. 2018;36(12):1260-1265. doi: 10.1200/JCO.2017.77.4893
- Advancing Education on Biosimilars Act of 2021, S 164, 117th Cong (2021-2022). Accessed May 19, 2022. www.congress.gov/bill/117th-congress/senate-bill/164
- Patel KB, Arantes Jr LH, Tang WY, Fung S. The role of biosimilars in value-based oncology care. Cancer Manag Res. 2018;10:4591-4602. doi: 10.2147/CMAR.S164201

