
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Unmet Clinical Needs in the Diagnosis and Management of Pulmonary Arterial Hypertension
Pulmonary hypertension (PH) is a complex, progressive, rare disease.1,2 PH is associated with considerable clinical burden and a high mortality rate. Pulmonary arterial hypertension (PAH) is a subtype of PH associated with high social, physical, and economic burdens. An estimated 500 to 1000 cases of PAH are diagnosed in the United States each year (2.3 cases per million Americans).3-5 Although PAH can develop at any age, it is most commonly diagnosed in patients who are aged 65 years or more and predominantly affects women.5-7 An estimated 30,000 people in the United States are living with PAH.8
Although some cases of PAH have known causes, such as genetic factors, many cases have no identifiable underlying cause and are designated as idiopathic. Early diagnosis and treatment are critical for improving patient outcomes, and patients should receive appropriate specialized care as soon as possible after diagnosis. Treatments are available for improving the symptoms and delaying the progression of PAH, but the disease remains incurable. Various treatments that are currently undergoing phase 2 and 3 clinical trials show potential for overcoming the limitations of previous therapies.
Pathophysiology
Clinically, PH is described as high blood pressure that develops in the vessels between the lungs and heart, which leads to increased muscle development in the vessel walls.9 Vascular remodeling involving narrowing of the arteries traveling from the right ventricle of the heart to the lungs results in restricted flow, leading to increased blood pressure in these arteries. This excess workload on the heart can cause weakness and failure of the heart muscle.4 With fewer vessels available to transport oxygen, blood oxygen levels are decreased. When there is insufficient oxygenation, the kidneys release the vasoconstrictor renin to activate the renin–angiotensin–aldosterone system, through which a series of hormones is stimulated to eventually cause further systemic vasoconstriction and ultimately increase blood pressure.10
PH is divided into 5 subtypes according to the underlying associated heart or lung disease.7,11,12 Specifically, group 1 is represented by PAH. The etiology and pathophysiology of PAH are multifactorial and not well-understood but are known to involve the endothelin pathway, prostacyclin pathway, and nitric oxide pathway.13,14
Guidelines, Diagnosis, and Classification
The classification of PH based on the proceedings from the Sixth World Symposium on Pulmonary Hypertension were retained in the 2022 European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines. These proceedings resulted in the development of a treatment algorithm based on risk stratification of patients with PAH.15 The updated guidelines were endorsed by the International Society for Heart and Lung Transplantation and European Reference Network for rare respiratory diseases.7 The updated ESC/ERS guidelines outlining the criteria for classifying patients with PAH into 6 subgroups are shown in Table 1.7,12



Approximately 50% to 60% of all PAH cases are idiopathic,7 although several risk factors (eg, diet, environment, genetics) can contribute to the disease risk and severity.16-18 The BMPR2 gene is the most common and well-studied gene associated with PAH.7 Individuals with BMPR2 mutations have a 20% lifetime risk of developing PAH; women who carry the mutated gene have a 42% risk compared with a 14% risk in men.7
Diagnostic Approaches
Physical examination, laboratory assessments, and other diagnostic tools are used to determine the cause of PAH and potential associated comorbidities.7 Laboratory testing can reveal the levels of N-terminal pro-brain natriuretic peptide (NT-proBNP), which are increased in the plasma of patients with PAH and right ventricular (RV) dysfunction. A BNP level more than 2-fold higher than normal levels indicates an increased risk of developing PAH.19 Echocardiogram, chest x-ray, and spirometry are important tools that are used to make a preliminary diagnosis of PH. Ultimately, hemodynamic assessment by right heart catheterization (RHC) is required to confirm a PAH diagnosis.6
Inappropriate and Delayed Diagnosis
The diagnosis of PAH is complex because symptoms can be heterogenous and nonspecific. These symptoms include dyspnea upon exertion, chest pain, and fatigue. Most symptoms are related to progressive RV dysfunction.7 Patients with PAH present with dyspnea, chest pain, arrhythmias, hemoptysis, syncope, and signs of right-sided heart failure (eg, fluid retention, reduced renal blood flow, activation of the renin–angiotensin–aldosterone system).5-7,11,20 Symptoms of PAH are often observed for several years before an accurate diagnosis is reached; without appropriate treatment, this can result in disease progression.13 Misdiagnosis or delayed diagnosis of PAH is common and occurs in up to 85% of at-risk patients.20
Screening Tools
The 2022 ESC/ERS guidelines provide detailed risk stratification charts and subsequent treatment recommendations for PAH. These guidelines recommend annual echocardiography screening for PAH in asymptomatic first-degree relatives of patients with identified PAH-causing mutations (eg, BMPR2 mutations).7 Blood biomarkers (BNP, NT-pro-BNP, and serum urate) are also included in PAH screening algorithms.
The time from symptom onset to diagnosis is often more than 2 years, highlighting the need for improved screening for PAH, particularly because therapies may be more effective when initiated early.7 Despite the frequent use of health care services prior to diagnosis, there are many missed opportunities for screening. Also, further research is needed to identify surrogate blood biomarkers as a potential screening tool for PAH, which may reduce the time to diagnosis.21
A multidisciplinary team of cardiologists, pulmonologists, thoracic surgeons, and others must ensure that they are educated on PAH screening tools to improve outcomes.7 A single-center, retrospective study of patients with PAH (n = 580) was conducted using data from 2008 to 2018 to determine if management at specialty care centers (SCCs) is associated with improved outcomes.22 Study results showed that patients who received care at an SCC had significantly longer survival times and fewer hospital admissions compared with those not managed at an SCC (P < .001). In addition, medication use (vasodilator use [P < .001]) and disease monitoring (repeated pulmonary function testing and repeated transthoracic echocardiogram; P < .024 and P < .004, respectively) were significantly more frequent in patients treated at SCCs compared with the frequency in those not undergoing care at an SCC.22 The findings strongly support that health care providers should refer patients with PAH to SCCs as early as possible.
Differential diagnosis among PH groups is important to correctly diagnose PAH. Most patients with PH display abnormal findings on chest radiography; however, normal chest X-ray results do not exclude PH, including PAH.7 Echocardiography can be performed to screen for PAH in patients presenting with characteristic symptoms (eg, unexplained breathlessness) or to confirm a PAH diagnosis.7,21 Echocardiogram findings during RHC showing an elevated heart rate and RV dysfunction strongly indicate a diagnosis of PAH23; right axis deviation and RV hypertrophy also are common.11
Suspected cases of PAH can be distinguished from other forms of PH based on how patients respond to vasodilator challenge in vasoreactivity testing performed during RHC as part of the echocardiogram. A positive result is considered as a decrease in the pulmonary arterial pressure of at least 10 mmHg to a mean pulmonary arterial pressure of no more than 40 mmHg. The test results can provide insight that is useful for making treatment recommendations. This method can be used to identify patients who may respond to treatment with calcium channel blockers (CCBs). In a landmark study performed in 1992, Rich and colleagues selected patients who showed a positive acute vasodilator response (responders) to CCBs in vasoreactivity testing for further treatment with these blockers.24 The authors demonstrated that patients identified in testing and subsequently treated with CCBs showed significantly improved survival rates after 1 year compared with the survival rate of nonresponders (94% vs 55%, respectively; P = .003).24
Additionally, pulmonary function testing is important for distinguishing between PH groups, assessing comorbidities, and determining severity. These tests should include forced spirometry, body plethysmography, lung diffusion capacity for carbon dioxide, and analysis of arterial blood gas.7
Follow-up Assessments of Patients With PAH
Referring patients to specialized centers is essential to ensure that patients are provided with education, informed on behavioral modifications, and given access to social and psychological support.7 Regular risk assessment of patients diagnosed with PAH is recommended to achieve or maintain a low-risk status. For example, as part of pulmonary function testing,21 the 6-minute walk test (6MWT) is widely performed to evaluate the exercise capacity and predict the prognosis of patients with PAH.7,25 In a multinational, prospective registry (COMPERA) of newly diagnosed patients with PAH (n = 2391) receiving targeted medical therapy, cut-off values for the 6MWT were identified as prognostic factors for the risk of death and as clinically relevant and important therapeutic targets.25
Despite the availability of assessment tools, a survey of US providers in 2017 showed that after a PAH diagnosis, patient follow-up and the frequency of assessments (clinic visits, 6MWT, echocardiogram, BNP level measurement, and RHC) varied among providers and were not aligned with guideline recommendations.26 After a patient is diagnosed with PAH, echocardiography and cardiac MRI should be performed at baseline and every 3 to 6 months after changes in therapy, both in stable patients and in cases of clinical worsening, to assess whether the disease is stable or progressing.7
Classification
The clinical classification of PH has undergone several revisions over the last 50 years.12 Guidelines from the American College of Chest Physicians (CHEST) were updated in 2019 and are used in the United States to provide guidance for diagnosing and managing patients with PAH, and are generally in alignment with the ERS/ESC guidelines.7,19 The 2022 ESC/ERS guidelines state that PAH is characterized as a mean pulmonary artery pressure of at least 25 mmHg, pulmonary wedge pressure of no more than 15 mmHg, and pulmonary vascular resistance of at least 3 Wood units.6,11,27 According to most PAH registries, idiopathic PAH is the most common subtype (approximately 50%-60% of all cases); PAH associated with connective tissue disease, coronary heart disease, and portal hypertension comprises the remaining cases.7
Although therapies are available for managing PAH symptoms, they do not treat the underlying cause of the disease.7 Various PAH-specific treatments, particularly combination therapies, have been shown to delay disease progression.14 However, currently available PAH therapies cannot reverse vascular remodeling; thus, although delayed, the disease still progresses.28
Advances are needed to support the early identification and diagnosis of patients with PAH. Additionally, tools for risk assessment to predict disease worsening should be developed. These approaches will allow for earlier treatment initiation with safe and effective options that prevent PAH progression and improve patient outcomes compared with those obtained using current approaches.28
Burden of Disease
PAH remains a deadly disease and is associated with various burdens on patients and their families. The clinical burden, comorbidities, economic impact, and quality of life (QOL) impact of PAH are discussed below.
Clinical Burden
PAH remains a deadly disease with high mortality rates. According to analysis of the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL), the 1-, 2-, and 3-year mortality rates of patients with PAH are 10%, 19%, and 25%, respectively.29 Chang et al stratified patients using various validated risk scores and found that the 3-year mortality rates of patients stratified into the low-, intermediate-, and high-risk groups were 7% to 11%, 18% to 20%, and 28% to 55%, respectively. Thus, results show that patients with high-risk disease have low survival rates after 3 years. The 1-year mortality rate of patients treated with a combination of therapies was lower than that in patients treated with monotherapy (7% vs 13%, respectively), although there was no difference between groups at 2 and 3 years.29
Comorbidities
PAH often occurs simultaneously with other conditions that have similar symptoms, which can complicate the identification of PAH and lead to diagnostic delays. Additionally, comorbid conditions that increase pulmonary artery pressure and cardiac output can promote PAH progression. PAH is associated with various conditions such as connective tissue disease (including systemic sclerosis and lupus), HIV infection, coronary heart disease, portal hypertension, and congenital abnormalities.7,30 Prevalence of comorbidities is higher in older patients compared with younger patients. Hjalmarsson and colleagues found that among patients with PAH, 22% of older patients (≥ 65 years) had at least 4 comorbid conditions vs 4% of younger patients (≤ 65 years).31 Older patients tend to show increased rates of ischemic heart disease, coronary artery disease, hypertension, atrial fibrillation, diabetes, and hypothyroidism.32 Lower survival rates may be related to the number of comorbidities. However, the risk factors for PAH are not independent from those of other conditions, and therefore some treatments may reduce the severity of comorbid diseases.
There is no cure for PAH, and many patients experience an aggressive disease course and poor survival outcomes.33 According to an analysis of data from the REVEAL registry (n = 2635; up to February 2013), the 5- and 7-year survival rates of patients with PAH are 57% and 49%, respectively, despite treatment with PAH-specific therapies.34
Economic Impact
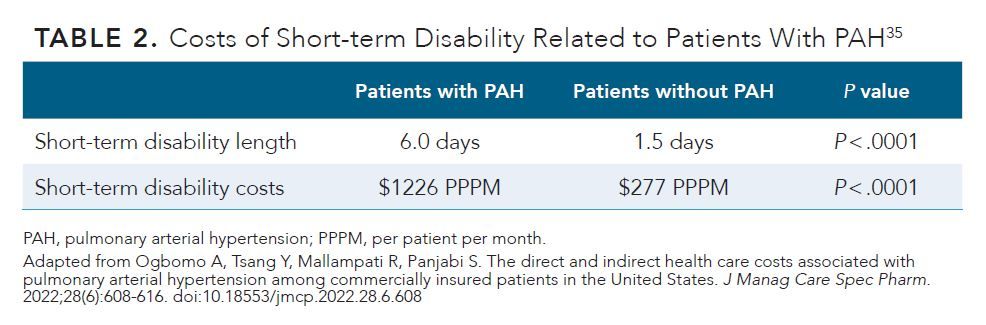
The high economic impact of PAH has also been widely recognized. Because of its high morbidity and mortality, PAH-related hospitalizations are a driving factor in health care costs. Drivers of indirect costs related to PAH include productivity losses and short-term disability expenses, which total in the thousands of dollars per patient per month (PPPM) (2018 US$) (Table 2).35 In a retrospective analysis of the economic burden of PAH using the MarketScan Health and Productivity Management Database, patients with PAH (age, 18-64 years) were defined as incident (diagnosed ≤ 6 months before enrolling in the study; n = 1293) or prevalent (diagnosed > 6 months before enrolling in the study; n = 455).35 Control participants matched by age, sex, and region were also evaluated for comparison. Both prevalent and incident cases were associated with more inpatient stays PPPM compared with those of controls (0.07 days for prevalent patients and 0.10 days for incident patients vs 0.00 days in the control group) (P < .001 for both comparisons). The length of hospital stays PPPM showed the same trend (0.63 days for prevalent cases vs 0.02 days in the control group; 0.92 days for incident patients vs 0.01 days in the control group; P < .001 for both comparisons). Mean all-cause health care costs were significantly higher in the prevalent ($9915 vs $359 PPPM; P < .001) and incident ($9353 vs $336 PPPM, P < .0001) populations compared with those in the control group. Key drivers of expenditures in the prevalent patient population were inpatient (46.6%) and pharmacy (28.0%) costs, whereas inpatient costs (66.0%) were the largest contributor to expenditures in the incident population.35


Quality of Life
Worsening PAH symptoms, such as dyspnea, fatigue, and a lack of energy, can lead to functional impairments. Patients with PAH have difficulty completing everyday tasks, including climbing stairs, doing housework, and shopping for household needs.32 The health of patients can be evaluated using the 36-item Short Form Health Survey (SF-36), which is used to query patients on 8 categories including vitality, physical functioning, physical role functioning, emotional role functioning, social role functioning, and mental health. A higher score on the SF-36 indicates a higher QOL. Patients reported a low QOL according to the results of the SF-36 survey, specifically because of the high prevalence of physical pain, stress, sleep disorders, anxiety, and depression.30 The caregivers of patients with PAH experience similar QOL impairments.32 Losses of productivity because of the inability to work full-time (or work at all) affect patients, families, employers, and payers.35
Conclusions
PAH is a rare but serious progressive condition with a high mortality rate. There are currently no curative treatments; although PAH-specific treatments help delay progression, these medications mostly address the disease symptoms and not the underlying pathophysiology. There are several unmet needs in the management of PAH, including the need for earlier diagnosis, more frequent evaluations once diagnosed, improved adherence to treatment along with referrals to specialized treatment centers, and drugs that target the underlying disease mechanisms. Furthermore, pharmacologic therapy for PAH requires a lifelong commitment, along with costly and frequent monitoring.36 Improvements in drugs that target the specific disease mechanisms with further enhanced disease-stabilizing effects are urgently needed.
References
- Beshay S, Sahay S, Humbert M. Evaluation and management of pulmonary arterial hypertension. Respir Med. 2020;171:106099. doi:10.1016/j.rmed.2020.106099
- Emmons-Bell S, Johnson C, Boon-Dooley A, et al. Prevalence, incidence, and survival of pulmonary arterial hypertension: a systematic review for the global burden of disease 2020 study. Pulm Circ. 2022;12(1):e12020. doi:10.1002/pul2.12020
- National Organization for Rare Disorders. Pulmonary arterial hypertension. Updated March 2, 2021. Accessed January 17, 2023. https://rarediseases.org/rare-diseases/pulmonary-arterial-hypertension/?filter=Affected+Populations
- Learn about pulmonary arterial hypertension. American Lung Association. Updated January 10, 2023. Accessed January 17, 2023. https://www.lung.org/lung-health-diseases/lung-disease-lookup/pulmonary-arterial-hypertension/learn-about-pulmonary-arterial-hypertension
- Frost AE, Badesch DB, Barst RJ, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US contemporary registries. Chest. 2011;139(1):128-137. doi:10.1378/chest.10-0075
- Dodson MW, Brown LM, Elliott CG. Pulmonary arterial hypertension. Heart Fail Clin. 2018;14(3):255-269. doi:10.1016/j.hfc.2018.02.003
- Humbert M, Kovacs G, Hoeper MM, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: developed by the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Heart J. 2022;43(38):3618-3731. doi:10.1093/eurheartj/ehac237
- Williamson T. The latest in diagnosis and care for pulmonary arterial hypertension. 2023. Accessed March 3, 2023. https://www.kansashealthsystem.com/news-room/blog/0001/01/pulmonary-arterial-hypertension-care
- Pulmonary hypertension. Centers for Disease Control. Updated December 3, 2019. Accessed January 17, 2023. https://www.cdc.gov/heartdisease/pulmonary_hypertension.htm
- Maron BA, Leopold JA. The role of the renin-angiotensin-aldosterone system in the pathophysiology of pulmonary arterial hypertension (2013 Grover Conference Series). Pulm Circ. 2014;4(2):200-210. doi:10.1086/675984
- Ruopp NF, Cockrill BA. Diagnosis and treatment of pulmonary arterial hypertension: a review. JAMA. 2022;327(14):1379-1391. doi:10.1001/jama.2022.4402
- Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S43-S54. doi:10.1016/j.jacc.2009.04.012
- Levine DJ. Pulmonary arterial hypertension: updates in epidemiology and evaluation of patients. Am J Manag Care. 2021;27(suppl 3):S35-S41. doi:10.37765/ajmc.2021.88609
- Sitbon O, Jaïs X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Resp J. 2014;43(6):1691-1697. doi:10.1183/09031936.00116313
- Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J.2019;53(1):1801889. doi:10.1183/13993003.01889-2018
- Rhodes CJ, Batai K, Bleda M, et al. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med. 2019;7(3):227-238. https://doi.org/10.1016/S2213-2600(18)30409-0
- Callejo M, Barberá JA, Duarte J, Perez-Vizcaino F. Impact of nutrition on pulmonary arterial hypertension. Nutrients. 2020;12(1):169. doi:10.3390/nu12010169
- Sydykov A, Mamazhakypov A, Maripov A, et al. Pulmonary hypertension in acute and chronic high altitude maladaptation disorders. Int J Environ Res Public Health. 2021;18(4):1692. doi:10.3390/ijerph18041692
- Klinger JR, Elliott CG, Levine DJ, et al. Therapy for pulmonary arterial hypertension in adults: update of the CHEST Guideline Expert Panel Report. Chest. 2019;153(3):565-586. doi:10.1016/j.chest.2018.11.030
- Maron BA, Galiè N. Diagnosis, treatment, and clinical management of pulmonary arterial hypertension in the contemporary era: a review. JAMA Cardiol. 2016;1(9):1056-1065. doi:10.1001/jamacardio.2016.4471
- Kiely DG, Lawrie A, Humbert M. Screening strategies for pulmonary arterial hypertension. Eur Heart J Suppl. 2019;21(suppl K):K9-K20. doi:10.1093/eurheartj/suz204
- Pi H, Kosanovich CM, Handen A, et al. Outcomes of pulmonary arterial hypertension are improved in a specialty care center. Chest. 2020;158(1):330-340. doi:10.1016/j.chest.2020.01.046
- Bonderman D, Wexberg P, Martischnig AM, et al. A noninvasive algorithm to exclude pre-capillary pulmonary hypertension. Eur Respir J. 2011;37(5):1096-1103. doi:10.1183/09031936.00089610
- Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76-81. doi:10.1056/NEJM199207093270203
- Zelniker TA, Huscher D, Vonk-Noordegraaf A, et al. The 6MWT as a prognostic tool in pulmonary arterial hypertension: results from the COMPERA registry. Clin Res Cardiol. 2018;107(6):460-470. doi:10.1007/s00392-018-1207-5
- Kingrey JF, Zhou CY, Dalal B, Elwing JM. Expert provider survey of longitudinal assessment in patients with pulmonary arterial hypertension. Heart Lung. 2023;58:34-38. doi:10.1016/j.hrtlng.2022.10.016
- Prins KW, Thenappan T. World Health Organization group I pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34(3):363-374. doi:10.1016/j.ccl.2016.04.001
- GlobalData Healthcare. PAH late-stage pipeline shows potential to expand existing therapy pathways. Pharmaceutical Technology. October 14, 2020. Accessed March 22, 2023. https://www.pharmaceutical-technology.com/comment/pah-market-therapy-pathways
- Chang KY, Duval S, Badesch DB, et al; PHAR Investigators. Mortality in pulmonary arterial hypertension in the modern era: early insights from the Pulmonary Hypertension Association Registry. J Am Heart Assoc. 2022;11(9):e024969. doi:10.1161/JAHA.121.024969
- Sarzyńska K, Świątoniowska-Lonc N, Dudek K, et al. Quality of life of patients with pulmonary arterial hypertension: a meta-analysis. Eur Rev Med Pharmacol Sci. 2021;25(15):4983-4998. doi:10.26355/eurrev_202108_26455
- Hjalmarsson C, Rådegran G, Kylhammar D, et al. Impact of age and comorbidity on risk stratification in idiopathic pulmonary arterial hypertension. Eur Respir J. 2018;51(5):1702310. doi:10.1183/13993003.02310-2017
- Delcroix M, Howard L. Pulmonary arterial hypertension: the burden of disease and impact on quality of life. Eur Respir Rev. 2015;24(138):621-629. doi:10.1183/16000617.0063-2015
- Gale S. The evolving treatment landscape of pulmonary arterial hypertension. Am J Manag Care. 2021;27(suppl 3):S42-S52. doi:10.37765/ajmc.2021.88610
- Benza RL, Kanwar MK, Raina A, et al. Development and validation of an abridged version of the REVEAL 2.0 Risk Score Calculator, REVEAL Lite 2, for use in patients with pulmonary arterial hypertension. Chest. 2021;159(1):337-346. doi:10.1016/j.chest.2020.08.2069
- Ogbomo A, Tsang Y, Mallampati R, Panjabi S. The direct and indirect health care costs associated with pulmonary arterial hypertension among commercially insured patients in the United States. J Manag Care Spec Pharm. 2022;28(6):608-616. doi:10.18553/jmcp.2022.28.6.608
- Schikowski EM, Swabe G, Chan SY, Magnani JW. Association between copayment and adherence to medications for pulmonary arterial hypertension. J Am Heart Assoc. 2022;11(22):e026620. doi:10.1161/JAHA.122.026620

