
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
The Role of Managed Care Professionals in Improving Care for Patients With ALS
Abstract
Amyotrophic lateral sclerosis (ALS) is a debilitating disease of progressively worsening physical function. The direct medical, direct nonmedical, and indirect costs of care for these patients is significant, and annual costs can be around $70,000 on average. Major drivers of cost can include hospitalizations, in-home care, nutrition, physical and occupational therapy, and medication interventions. Beyond the monetary costs, patients also endure a decreased quality of life, which may also burden caregivers and family members. Appropriate utilization management by managed care organizations and support for the multidisciplinary care team approach can help to ensure appropriate care, with a goal of optimizing clinical outcomes and potentially delaying disease progression. Although ALS has no cure, progressive gene therapy is currently being investigated and may offer promise. Thus, gene therapy may necessitate the need for novel and creative approaches to cost-management strategies in the future.
Am J Manag Care. 2020;26:S198-S205. https://doi.org/10.37765/ajmc.2020.88484
Economic Burden of Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) represents a significant economic burden in the United States, which may or may not be realized by managed care organizations. A moderate estimate in 2010 suggested the total economic burden of ALS was just over $1 billion. Although this number may not seem large when compared with total healthcare expenditures, it is important to note that the overall prevalence was about 20,000 at the time, with an annual incidence rate of 5000.1-3 This incidence rate is about 2 cases for every 1,000,000 people and is increasing each year.4 If ALS were to reach incidence numbers similar to other chronic diseases, the total economic burden would be much higher and more significant. Disease characteristics that may affect costs of care include age of onset (40-75 years old) and average time of survival after diagnosis, which is currently just 2 to 5 years, with 20% of patients with ALS living longer than 5 years, 10% living 10 years, and 5% living 20 or more years.1-5 Along with the increasing incidence rate per year, physicians have noted an increasing number of younger patients with ALS.4 Unfortunately, this disease has no known cure and shows a rapid and progressively worsening disease state. As disability scores increase, so do costs associated with overall care.1
Current disease-modifying medication therapy focuses on the delay of disease progression. Other treatments are available to provide supportive care for sequelae of disease that can include cognitive and behavior changes, pseudobulbar affect (PBA, defined as sudden outbursts of laughter and/or crying), difficulty communicating, spasticity, pain, nutrition, and mobility.6
When considering the total cost of care for ALS, it is important to include not only the cost of medications but also costs under the medical benefit, such as institutional and ambulatory services, professional services, durable medical equipment (DME), and other ancillary services. In one retrospective claims analysis conducted from 2008 to 2015 of the Truven Health Analytics MarketScan Database, the top 3 drivers of ALS costs were inpatient care, surgery, and prescriptions. One consideration is this was before the FDA approval of edaravone in 2017, which would have increased prescription drug costs. In this same study, the estimated total cost of care per member per month (PMPM) was $3460 compared with $486 for an average adult patient aged 16 years and older.7 One limitation with the current studies evaluating the cost of ALS is the limited amount of patient data. Another annual cost estimate for the total cost of care in 2015 was around $70,000, which is approximately $5833 PMPM.3 However, it should be noted the cost of care will vary greatly between patients due to heterogeneity of patient treatment plans.
In a case study of 1 patient with ALS, cost data were collected over 10 years (2001-2010). The total cost of care over 10 years was $1,433,992, of which 85% was paid by third-party payers, 9% paid by the patient, and 6% paid by charities. For this patient, the 3 main drivers of total costs were in-home care (46.7%), ventilation (14.8%), and in-hospital expenses (8.0%). Medications accounted for just 1.7% of the total cost of care. It bears noting, however, that this patient did not take riluzole. Use of riluzole and/or edaravone (approved after this case study) would have substantially increased the cost of medications in this case study.8
The American Academy of Neurology (AAN) developed quality measures in 1999 to address gaps in care and ensure proper utilization of evidence-based therapies for patients with ALS, which were updated in 2009.9,10 A summary of these quality measures is outlined in Table 1.9,10


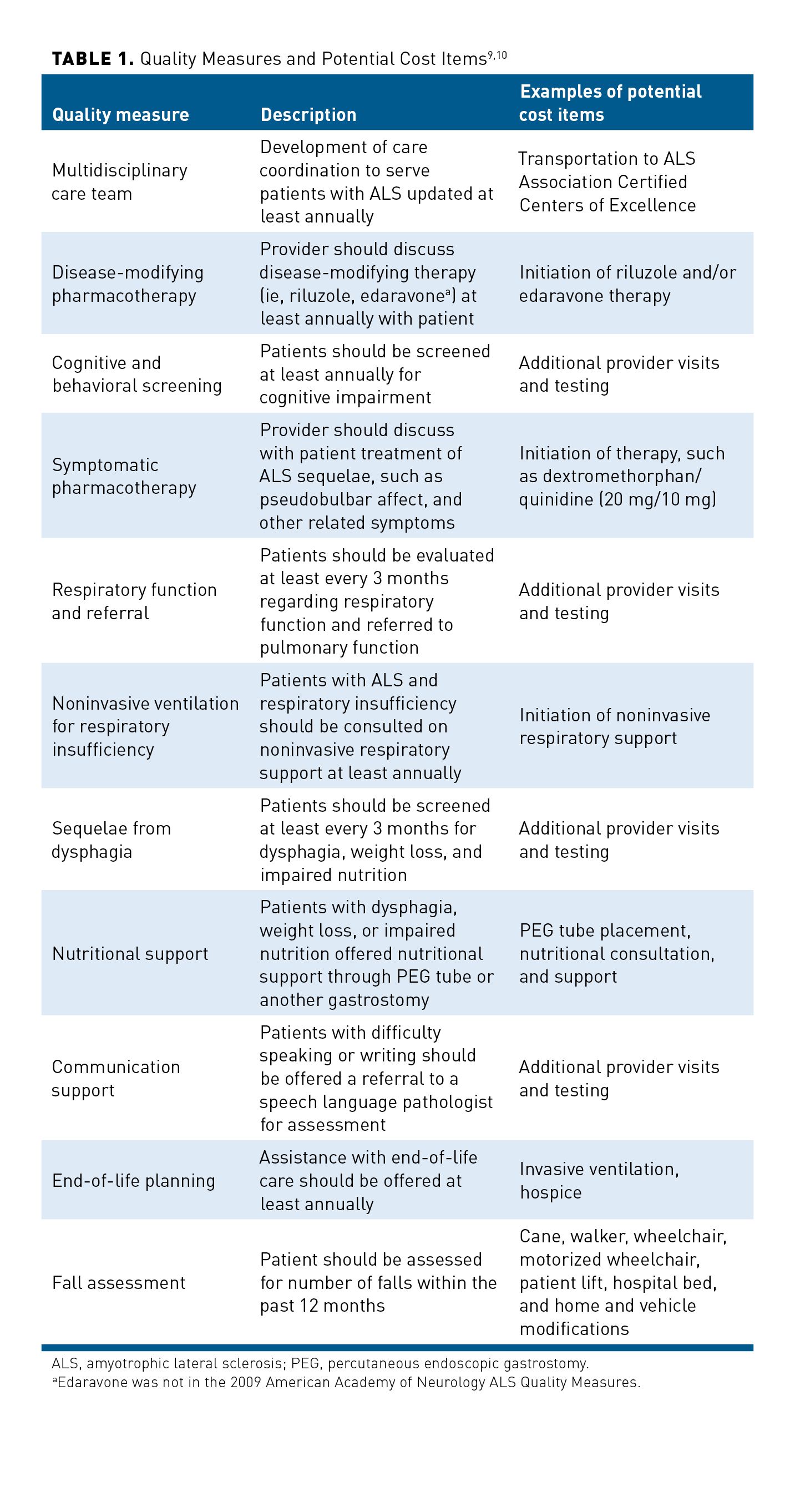
The following sections will review the direct medical costs (eg, healthcare utilization, physical therapy, pharmacotherapy), direct nonmedical costs (eg, DME, vehicle and home modifications), and indirect costs (eg, quality of life, caregiver and family burden). Each patient has a unique clinical presentation with a variable rate of disease progression as well as a different perspective on what quality of life means.11 These factors would drastically alter the total cost of care for each patient. Therefore, estimations of total cost of care will vary greatly between each patient.
Direct Medical Costs
Healthcare utilization costs include provider visits, hospitalizations, long-term care, nutrition, pharmacotherapy, and hospice.12-18 For a patient with ALS, hospitalizations can be a result of loss of respiratory function, pneumonia, nutrition, or other consequences of decreased physical functioning. In a 2010 study of Medicare patients with ALS, estimated costs were $13,324 for inpatient care and $10,745 for outpatient care (eg, physician office visits, occupational/physical therapy).1 Appropriate care coordination and multidisciplinary care teams may help to optimize care to decrease unnecessary medical costs and hospitalizations. Managed care organizations need to recognize that current disease-modifying treatment options are not cures, and the goal of treatment is to delay or slow disease progression. Thus, disease progression is inevitable and seeking inpatient care may not be avoidable.
Besides direct hospitalization costs, treatment of sequelae of ALS progression may also require treatment. For example, PBA is a known sequela of ALS. This condition is characterized by uncontrollable laughter and crying and may affect the patient’s quality of life. Treatment for PBA is limited as there is only 1 FDA-approved medication indicated for the treatment of symptoms. Dextromethorphan hydrobromide/quinidine sulfate (20 mg/10 mg) capsule has been shown in clinical trials to significantly decrease the number of laughing and crying episodes from baseline. Managed care organizations must recognize that this medication is indicated for any patient with PBA, which includes ALS and other mild cognitive impairment conditions.19
Pain and spasticity are common as ALS progresses, developing as secondary complications of musculoskeletal dysfunction due to limited mobility or loss of range of motion. Many patients with ALS report pain even in the early stages of their disease. Spasticity may compromise gait and manual dexterity and may lead to the development of painful joint contractures. The treatment of associated pain and spasticity is multimodal and includes nonpharmacologic (eg, massage, stretching, positioning, use of appropriate bed/chair/lift) and pharmacologic therapies (eg, muscle relaxants, laxatives, benzodiazepines). Risks of pharmacologic therapy should be weighed against potential benefits. Consultation and referral to physical therapy may offer patients a higher quality of life without the added adverse reactions, such as sedation. Increased sedation in patients with ALS may potentially increase risk of respiratory insufficiency.20 Decreased motor function can also lead to reduced ability or complete inability to eat, which can lead to malnutrition or need of enteral nutrition in later stages of the disease.11
Nutritional needs for patients are a significant contributor to total cost of care. In 2001, it was estimated that alternative nutritional maintenance was about $7300 per year. This does not include other ancillary needs or complications of enteral nutrition (eg, PEG surgery), and overall cost is undoubtably higher today than it was in 2001.14
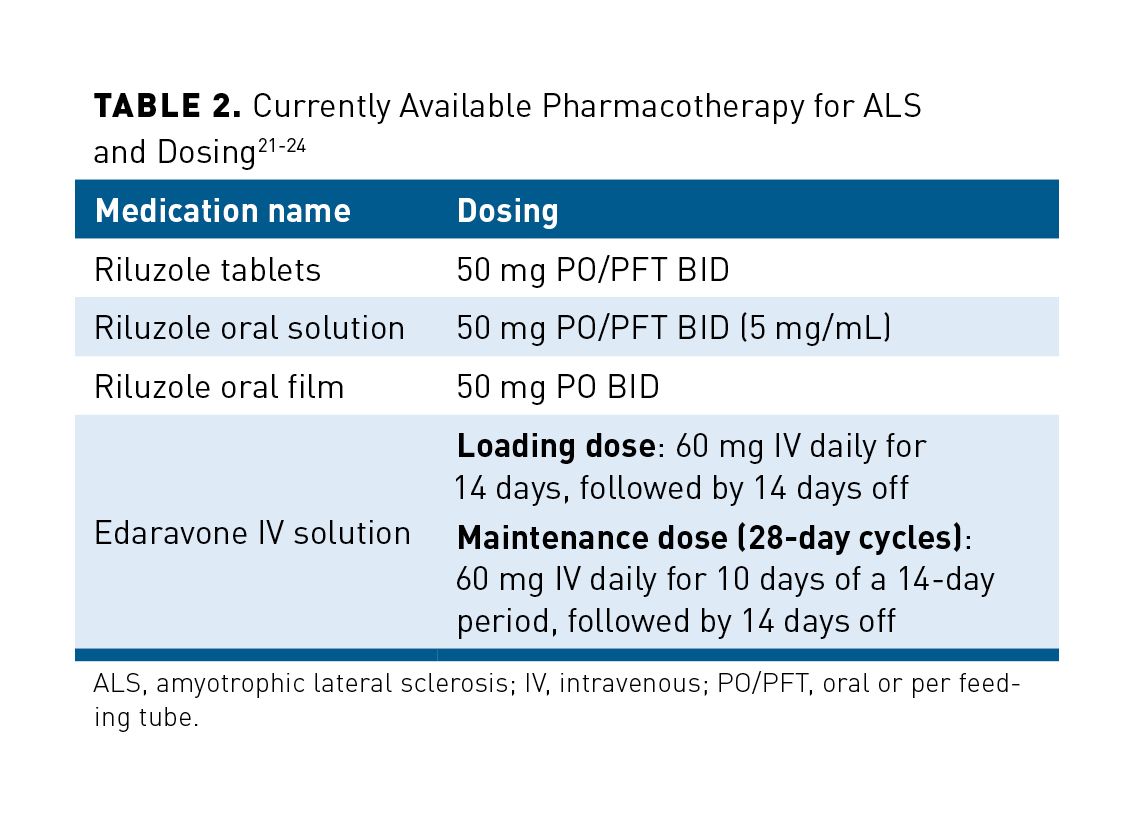
Although there are other direct medical costs that can be considered, one other significant cost associated with care is pharmaceutical treatment options. Current disease-modifying pharmacotherapeutic options for management of ALS are riluzole and edaravone. As previously noted, these treatment options neither reverse disease progression nor represent a cure, but they may help to slow disease progression. For more than 2 decades, riluzole oral tablets were the only disease-modifying therapy available for ALS. Riluzole is currently available as a generic tablet and a brand only oral solution. Most recently, an oral film formulation has been FDA approved for the treatment of ALS, but has not yet been launched into the market. Edaravone was approved for the treatment of ALS in 2017.3,21-25
Table 221-24 outlines the currently approved FDA pharmacotherapy options for ALS. It must be noted that edaravone starts with a loading dose of 60 mg daily for 14 days, followed by 14 days off. This is followed by 60 mg daily for 10 days of a 14-day period, followed by 14 days off.18,24


Direct Nonmedical Costs
Direct nonmedical costs include costs paid by payers or the patient that are not directly related to medical services or care. This includes DME, such as hospital beds, walkers, wheelchairs, and ventilation.26-30 Less obvious costs include patient lifts and electronic eye gaze devices that allow speech through a patient’s eye movements.13,31
There appears to be a direct correlation to the staging of the disease and the amount of direct nonmedical costs. Although there are multiple proposed staging methodologies for ALS, there is no accepted gold standard.5,27,32,33 One system that is suggested is outlined here:
Stage 1: Symptom onset involvement of 1 region (defined by origin of onset: bulbar, upper limb, lower limb, diaphragmatic region)
Stage 2: Symptomatic involvement of a second region
Stage 3: Symptomatic involvement of a third region
Stage 4: Nutritional or respiratory failure (terminal)
Stage 5: Death
The disease stage can also be defined by the amount of aid needed to ambulate, such as moving from a cane or walker to a wheelchair or moving from noninvasive ventilation (NIV) to invasive mechanical ventilation.5,30 In one retrospective study of Centers for Medicare & Medicaid Services (CMS) Limited Data Set, patients were evaluated on the cost and time from ALS diagnosis to each milestone. A total of 1687 patients who had a diagnosis of ALS for at least 6 months prediagnosis were identified. These milestones (M1-M6) as well as the average time (months) and cost of ALS treatment from diagnosis to milestones are summarized in Table 3.34


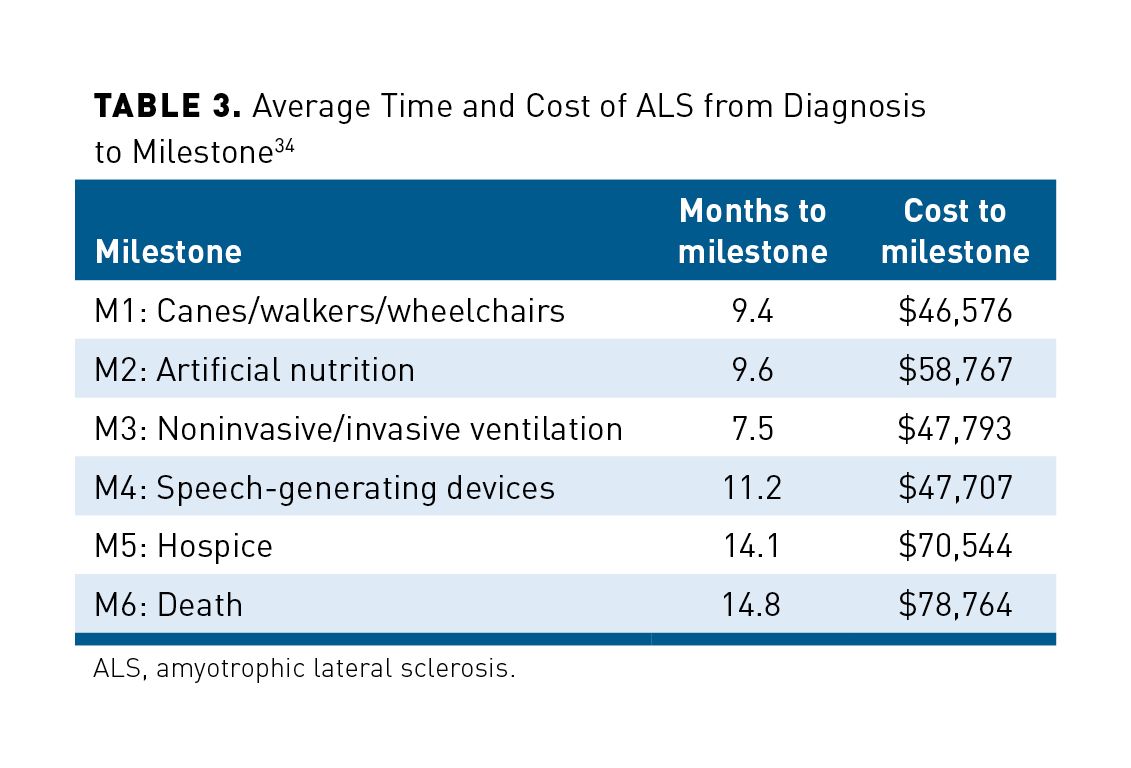
The study also found that for patients with at least 12 months postdiagnosis data (n = 956) the average total cost of treatment was higher in patients with ALS compared with a matched control group ($34,475 vs $32,165; P <.01). Compared with the matched control group, patients with ALS also had higher utilization of healthcare resources, which included physician office visits (96.5% vs 91%), outpatient hospital visits (91.3% vs 80.1%), and at least 1 DME claim (65.4% vs 52.4%; all P <.01). Study results showed decline in physical function is progressive, and milestones may occur concurrently or in rapid succession.34
The origin of ALS symptoms, limb versus bulbar, may play a role in how quickly a patient progresses. In one study, patients with ALS that originated in the lower extremities (limb onset) progressed to walking aids at a faster rate compared with bulbar (head/neck) or upper extremity onset. Those who required a PEG tube sooner more likely had a bulbar onset of ALS symptoms. However, this study found that regardless of where the initial onset of symptoms occurred, many patients required the full array of DME (ie, cane, walker, PEG, wheelchair, NIV, ankle-foot orthosis) and nonmedical interventions for 60% to 80% of the entirety of their ALS disease duration. The study authors concluded that these DME and interventions will likely be needed in the second half of ALS progression, which averages about 15 months after diagnosis.30 The highest cost noted was a customized power wheelchair, which in 2008 was estimated to be about $27,800.26,30 Other significant costs were PEG tube placement ($2000), NIV machine ($3000), and equipment installed in the home, such as lift chair ($900), stair lift ($2000), and vertical lift ($4000).30
In terminal patients, invasive ventilation may be warranted instead of NIV.29 NIV can be provided through a mask by a bi-level intermittent positive airway pressure (BiPAP) machine; invasive ventilation is given through a tracheostomy.1,14 Both noninvasive and invasive ventilation have been shown to prolong life in patients with ALS, but there is a significant cost incurred with invasive ventilation. Therapy that may delay time to tracheostomy or disease progression (ie, riluzole, edaravone) represents an opportunity for potential cost-savings. In one case study, total cost of ventilation over 10 years for a patient was $212,430. Other intangible costs that may be incurred by patients are costs of back-up batteries and generators in case of a power outage and increased utilization of in-home care to ensure around-the-clock monitoring for respiratory function.8 Other apparatus that may be required by some patients who have lost the ability to communicate verbally or in writing are augmented communication devices. These can range from $38 for a visual eye blink board to more than $30,000 for an ALS-specific computer system to track eye movement.13
Indirect Costs: Quality of Life and Caregiver and Family Burden
ALS is not only a disease of healthcare resource utilization costs but also of significant cost not related to treatment. There are significant impacts upon the loss of productivity, work, and quality of life for patients as well as the burden on their immediate family and caregivers.1,5,8,16,35
Caregivers have described their first-hand experiences of providing care to patients with ALS, and the concept of First Half or Second Half method of communicating, which refers to the first or second half of the alphabet. As described earlier, many patients in the advance stages of ALS lose the ability to communicate, and their only way to do so is through their eye movements. To spell out words, each letter would have to be painstakingly spelled out by gesturing with their eyes. To save time and effort, it is easier to split the alphabet in half. Caregivers also talk about having to lubricate and close their patient’s eyes at night so they do not dry out, constantly suctioning fluid out of the tracheostomy, worrying if the ventilator battery will last if there is a power outage, and considering the ethical dilemmas of death with dignity and religious beliefs.
Calculating loss of income from work is difficult to determine. Although patients with ALS will decline physically, mental acuity does not diminish. In one estimate, a family would have an average income loss of $19,217 per year, with a personal indirect cost of $14,682. This will vary depending on the amount of care needed as well as who provides care to that family member.1 Other costs besides loss of income include increased cost of in-home care, especially in patients with ventilator needs. The total annual cost for supporting a mechanically ventilated (invasive ventilation) patient can be $18,965 on average, whereas costs for those on NIV are around $5650 and costs for those with no ventilators may be $1653.1
Additional costs of care can include retrofitting a vehicle for a patient, providing for power wheelchair transportation, and making house renovations, such as bigger entryways, support tracks for patient lifts, accessible bathrooms, and specialized hospital beds to decrease the risk of bed sores and other sequelae of being bedridden. In previously reported cases, the cost of an accessible van was between $30,000 and $36,000.8
In addition to the indirect costs, ALS has a significant and profound psychosocial impact on patients and caregivers.1,16,27,35,36 Increased disability and decreased quality of life associated with advanced stages of ALS have been correlated to worsening depression and anxiety scores.27 This intangible human cost is difficult to quantify; however, it is arguably one of the largest costs ALS imposes on a patient and the patient’s family. With ALS, it is inevitable that a patient has to face their mortality. Where it is legal, death with dignity laws have allowed patients with certain diseases the right to end their own life. It is not a decision taken lightly and has many legal procedures that must occur prior to implementation. However, for patients with ALS, death with dignity is a choice that many explore. In one study, patients with ALS in the state of Washington had the highest proportion of death with dignity compared with other diagnoses.36 With this decision comes many other dilemmas, such as conflicts with religious beliefs and family member wishes. As a note for managed care pharmacists, federal funding may not be used for services or medications under death with dignity.37
Utilization Management Considerations
Goal of Therapy
Both riluzole and edaravone are disease-modifying medications that do not result in a cure or reversal of disease progression. They are 2 different compounds with a mechanism of action in ALS that is still not well defined. The inclusion criteria and primary end points evaluated in pivotal trials also differed between riluzole and edaravone, though both agents were FDA approved for all patients with ALS. In the pivotal trials, riluzole significantly reduced the median time to tracheostomy or death (primary outcome) by 60 to 90 days in patients with ALS. However, there was no significant difference in muscle strength and neurological function.21
On the other hand, edaravone was studied in a slightly different population and with a different primary outcome. In pivotal trials, patients must have 2 years or fewer of a definite or probable diagnosis of ALS based on the El Escorial revised criteria, normal respiratory function (forced vital capacity ≥80%), and mostly retained functionality of daily living as defined by a score of 2 or more on all components of the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R), meaning these patients were early in their ALS disease course. It is important to note that at the time of study start, 90% of patients were on riluzole and could continue riluzole. The primary outcome in this study was the change (negative change indicating decreased functionality) in ALSFRS-R from baseline to week 24. At week 24, compared with placebo, patients on edaravone had significantly higher (though still decreased) ALSFRS-R scores (–5.01 vs –7.50), representing a 33% less worsening of function.27 Many have debated on what change in ALSFRS-R is considered clinically meaningful. In one survey of ALS clinicians, a decrease in ALSFRS-R score of 4 was considered clinically meaningful.38 Although edaravone decreased amount of functional decline, it did not stop or reverse the course of the disease through a 1-year follow-up.
Medical versus Pharmacy Benefit
Patients with ALS are qualified for Medicare benefits as soon as Social Security Disability Insurance payments start, which is approximately 5 months after a patient is deemed disabled.39 The difficulty for most patients is that prior to an official diagnosis of ALS there may be several months of differential diagnostic testing and evaluation that can increase both payer and patient costs.12
Medicare benefits include Part A (hospital), Part B (outpatient), Part C (Medicare Advantage), and Part D (prescription drug coverage). Medicare regulations require Part D formularies to have at least 2 drugs in each drug category or class (eg, U.S. Pharmacopeia, American Hospital Formulary Service, or custom). In most scenarios, riluzole will be managed under the Part D benefit, whereas edaravone will be covered under the Part B benefit because it is an intravenous infusion. However, there may be an instance where edaravone is given as a home infusion and billed as a Part D drug benefit.40 Each managed care organization is different, and internal policies should be followed in determining Part B or Part D determinations; however, there are opportunities for managed care organizations to potentially save money by promoting administration in a home setting as opposed to an institutional ambulatory setting. Part C Medicare Advantage plans will have the Part B versus Part D distinction for coverage as well as the possibility of integrated utilization management across the medical and pharmacy benefits.
Formulary Considerations for Riluzole and Edaravone
Because the riluzole tablets have been on the market for decades and are generically available, most health plans will not feel the need to implement strict utilization management criteria. If a prior authorization is imposed, it may require only attestation of an ALS diagnosis. Riluzole is also available as a branded oral suspension (approved in 2018) and a film (approved in 2019).21-23 Though the riluzole film has been approved, it is currently not commercially available.25 Managed care organizations should consider limiting utilization of these branded formulations to patients who are unable to tolerate swallowing whole oral tablets. Authorization criteria should include documentation of a previous trial of generic riluzole tablets (step therapy) and attestation of the patient being unable to administer or tolerate oral tablets.
Edaravone, approved in 2017, is a novel pharmacologic agent that was first approved for use in Japan in 2001 to facilitate the recovery from cerebrovascular accidents. It was then studied in Japan in 2011 for ALS, resulting in an approval in 2015. Initial trials of edaravone in the general ALS population in the United States failed to show any benefit. However, further dissection of the general ALS population indicated a benefit when used early in patients with ALS, leading to the pivotal trial of edaravone in early-stage ALS and its FDA approval.
It is important to note that, to date, there have been no head-to-head trials comparing the safety and efficacy of riluzole with edaravone. Claims of superiority of one agent over the other cannot be made. In fact, most patients in the edaravone clinical trials were taking riluzole and were allowed to continue taking their riluzole. Because coverage will default to the medical benefit, medical policy and precertification process will be used to manage edaravone utilization. The level of enforcement of the medical policy and precertification process will differ with each managed care organization. Medication policy and precertification criteria will largely mirror inclusion criteria from the pivotal trial, including a definite or probable diagnosis of ALS of no more than 2 years and, for the most part, normal physical and respiratory function.24 This would exclude many current patients with ALS who are beyond 2 years or whose disease has progressed to a stage where physical and respiratory function parameters do not satisfy the pivotal trial inclusion criteria.
Best Practices
The ALS Association and the Muscular Dystrophy Association (MDA) are 2 national nonprofit organizations focused on ALS. Both the ALS Association and the MDA collaborate with physicians and researchers to certify clinics.41,42
There is limited evidence to suggest that multidisciplinary care can directly lower patient cost.43-45 One study did suggest that earlier referral to multidisciplinary care may decrease cost but may not decrease time to ALS diagnosis.44 Regardless, formation of a multidisciplinary team is still an accepted quality measure of the AAN. If possible, patients should seek either an ALS Association Certified Treatment Center of Excellence or a Recognized Treatment Center to ensure optimal care.9,45
There should be little disagreement that care for patients with ALS—especially in the late stages—can be complex. Managed care organizations need to embrace and support the use of a multidisciplinary approach, integrating their care coordination capability to ensure the most cost-effective use of resources. Managed care pharmacists should play an integral and proactive role in the treatment and management of these patients to ensure utilization of appropriate medications. As new therapies are developed, pharmacists may have to facilitate patient enrollment into clinical trials at ALS Association Certified Treatment Centers of Excellence.42
Future Considerations
In September 2019, the FDA released a guidance on the development of clinical trials for ALS. The guidance suggests that trials not exclude patients with ALS based on characteristics such as age or disease stage unless scientifically necessary. This is an attempt to include a broader patient population in ALS trials. In addition, the FDA suggests primary end points should focus on patient-oriented outcomes, such as physical function, respiratory function, and mortality. It also acknowledges the potential for gene therapy as an option for ALS.46
Biologic therapy and gene therapy are currently being explored for use in ALS.47-49 Gene therapies are still early in development, but the early research shows promise. Current gene therapy research is focused on patients with ALS with mutations in superoxide dismutase 1 (SOD1) or C9orF72 genes. These mutations may lead to types of familial ALS, which accounts for approximately 10% of the ALS population.47-49 The vector utilized to target these genes is derived from the adeno-associated virus (AAV), the same vector that is being used for a recently approved gene therapy for another motor neuron disease, spinal muscular atrophy (SMA), called onasemnogene abeparvovec-xioi (Zolgensma). This gene target, along with a viable vector derived from AAV, gives some promise to a potential cure for ALS.50
Clinically, a cure for ALS would be nothing short of miraculous, but it does create a difficult situation for managed care organizations. Onasemnogene abeparvovec-xioi has set the pricing precedent for gene therapies. There is a strong possibility that managed care organizations will again be faced with balancing the cost of care with the clinical outcome in an attempt to determine the overall cost-effectiveness proposition. One way to evaluate treatment options in ultrarare and catastrophic conditions is through the use of a cost-effectiveness threshold (CET), which is the maximum cost at which the treatment is deemed to be cost-effective and the cost is justified. The CET evaluation framework was developed and executed by the Institute for Clinical and Economic Review (ICER) and is utilized by many evaluating organizations globally.48 The ICER is an independent research organization that objectively reviews both clinical and financial aspects of medications and other healthcare tools. This organization is considered a leading monitor for drug pricing and evaluates how drugs should be priced to reflect the benefit.50
The CET is usually calculated through an incremental cost-effectiveness ratio, which is a ratio of difference in cost between 2 possible interventions, divided by the difference in the effect, which is calculated based upon the number of years life is extended by with a quality-of-life modifier. This cost-effectiveness ratio is usually presented as a dollar amount for quality-adjusted life-year (QALY) gained. For normal health conditions, the CET has been described as $50,000 to $150,000 per QALY. Although there is no definitive CET for ultrarare conditions, the consensus range is between $175,000 and $500,000 per QALY for ultraorphan drugs. For example, a recently approved drug for SMA, nusinersen (Spinraza), has a ratio of more than $375,000 per QALY gained. Although this does not pass the upper limit of the CET of $500,000 per QALY, it is still an expensive agent that could be debated as not cost-effective.48
In order to provide policymakers with a broad view of cost-effectiveness, the ICER included the cost per life-year-gained (LYG) in conjunction with the cost per QALY. The difference between QALY and LYG is that the QALY calculation includes a utility measurement of quality of life and duration of life extension. The cost per LYG approach values any life extension with no regard to the quality of life. Hence, in treatments for which the impact to quality of life is minimal but which are associated with a significant extension of life, the cost per LYG may appear more favorable compared with the cost per QALY.50
A pharmacoeconomic review of edaravone by the Canadian Agency for Drugs and Technologies in Health (CADTH) estimated that the incremental cost-effectiveness ratio for edaravone was $1,440,786 (CAD) for patients with mild ALS.51 Some have questioned whether the ICER evaluation framework is broad enough to take into account those parameters upon which a value cannot be placed. One article points out that the current cost-effective evaluation methodology is only useful when the information available to drive the treatment decision process is similar between the physician and the evaluation tool.52 In other words, is the evaluation tool including the same level of information in the evaluation as the physician is using when making a treatment decision? Unfortunately, this does not usually occur. The physician will usually have more clinical and psychosocial information available to make the treatment decision, as opposed to what the payer or the evaluation tool assumes or takes into account to determine the value of the treatment. The ability to place a cost value upon these real-world decision parameters simply does not exist. A second point the author makes is that although best practices can be identified and endorsed, there will be clinical presentations where the best therapeutic options may not be the best practice but rather be in the best interest of the patient.52 These points highlight that coverage criteria fit most patients but not all. Therefore, managed care organizations should take into consideration provider judgment when making utilization management decisions.
In an attempt to blunt the impact of these high-cost therapies, managed care organizations are actively seeking creative ways to provide coverage for these medications while not financially overburdening the entity that holds the financial risk. Manufacturers acknowledge that the costs of these curative treatments are not easily managed. In a pilot venture, the New Drug Development ParadIGmS (NEWDIGS), a Massachusetts Institute of Technology-based group, is exploring new payment methodologies. This collaboration comprises payers, manufacturers, and other stakeholders. One of the features being investigated is a performance-based annuity payment model, which spreads out payments to the manufacturer over time, similar to a traditional mortgage. One payment model suggests a mobility of payments, so if a patient switches health plans, the new health plan is responsible for continued payments to the manufacturer.53,54 Managed care organizations may need to be creative in the future of payment models for these ultrarare and expensive curative therapies, if approved.
Conclusions
Managed care organizations have the difficult task of balancing clinical outcomes with the cost of care. Therefore, it is incumbent upon the organization to responsibly implement utilization management strategies to ensure appropriate use for medication treatment options for ALS. Although the current treatment options will cross the line of pharmacy versus the medical benefits, coverage determinations should take into account the clinical presentation with respect to the total cost of care as well as any psychosocial parameters in an effort to delay disease progression. Managed care organizations need to be supportive of the use of multidisciplinary care teams providing care to and supporting patients with ALS as well as initiate pharmacotherapy interventions earlier in the disease course to potentially provide optimal benefit. As gene therapy continues to be developed, innovative cost-containment strategies should be explored to manage the cost of care for patients with ALS.
Author affiliation: Winston Wong, PharmD, is president of W-Squared Group, Longboat Key, FL.
Funding source: This activity is supported by an educational grant from Mitsubishi Tanabe Pharma America, Inc.
Author disclosure: Dr Wong has no relevant financial relationships with commercial interests to disclose.
Authorship information: Substantial contributions to the concept and design; supervision; and critical revision of the manuscript for important intellectual content.
Address correspondence to: wwong@wsquaredgroup.com
Medical writing and editorial support: Andrew M. Abe, PharmD
References
1. The Lewin Group. Cost of Amyotrophic Lateral Sclerosis, Muscular Dystrophy, and Spinal Muscular Atrophy in the United States. The Lewin Group; 2012. Accessed April 28, 2020. mda.org/sites/default/files/Cost_Illness_Report_0.pdf
2. Hulisz D. Amyotrophic lateral sclerosis: disease state overview. Am J Manag Care. 2018;24(15 suppl):S320-S326.
3. Santaniello B. ALS managed care considerations. Am J Manag Care. 2018;24(15 suppl):S336-S341.
4. ALS incidence. ALS Treatment. Accessed July 13, 2020. alstreatment.com/amyotrophic-lateral-sclerosis-incidence/
5. Oh J, An JW, Oh SI, et al. Socioeconomic costs of amyotrophic lateral sclerosis according
to staging system. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(3-4):202-208.
doi: 10.3109/21678421.2014.999791
5. Amyotrophic lateral sclerosis (ALS). Muscular Dystrophy Association. Accessed April 28, 2020. mda.org/disease/amyotrophic-lateral-sclerosis/signs-and-symptoms
7. Cohen B, Balcells C, Hotchkiss B, Aggarwal K, Karaa A. A retrospective analysis of health care utilization for patients with mitochondrial disease in the United States: 2008-2015. Orphanet J Rare Dis. 2018;13(1):210. doi: 10.1186/s13023-018-0949-5
8. Obermann M, Lyon M. Financial cost of amyotrophic lateral sclerosis: a case study. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(1-2):54-57. doi: 10.3109/21678421.2014.951946
9. Miller RG, Brooks BR, Swain-Eng RJ, et al. Quality improvement in neurology: amyotrophic lateral sclerosis quality measures. report of the Quality Measurement and Reporting Subcommittee of the American Academy of Neurology. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(3-4):165-168. doi: 10.3109/21678421.2013.875706
10. Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology [published correction appears in Neurology. 2009;73(24):2134] [published correction appears in Neurology. 2010;74(9):781]. Neurology. 2009;73(15):1218-1226. doi: 10.1212/WNL.0b013e3181bc0141
11. Paganoni S, Karam C, Joyce N, Bedlack R, Carter GT. Comprehensive rehabilitative care across the spectrum of amyotrophic lateral sclerosis. NeuroRehabilitation. 2015;37(1):53-68. doi: 10.3233/NRE-151240
12. Schultz J. Disease-modifying treatment of amyotrophic lateral sclerosis. Am J Manag Care. 2018;24(15 suppl):S327-S335.
13. Fiorentino G, Esquinas AM. Cost-effectiveness associated with amyotrophic lateral sclerosis: some questions and answers pending. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(3-4):315-316. doi: 10.1080/21678421.2017.1408126
14. Ginsberg G, Lowe S. Cost effectiveness of treatments for amyotrophic lateral sclerosis: a review of the literature. Pharmacoeconomics. 2002;20(6):367-387. doi: 10.2165/00019053-200220060-00002
15. Ashworth NL, Satkunam LE, Deforge D. Treatment for spasticity in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2012;(2):CD004156. doi: 10.1002/14651858.CD004156.pub4
16. Burke T, Galvin M, Pinto-Grau M, et al. Caregivers of patients with amyotrophic lateral sclerosis: investigating quality of life, caregiver burden, service engagement, and patient survival. J Neurol. 2017;264(5):898-904. doi: 10.1007/s00415-017-8448-5
17. Gladman M, Dharamshi C, Zinman L. Economic burden of amyotrophic lateral sclerosis: a Canadian study of out-of-pocket expenses. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5-6):426-432. doi: 10.3109/21678421.2014.932382
18. Meng L, Bian A, Jordan S, Wolff A, Shefner JM, Andrews J. Profile of medical care costs in patients with amyotrophic lateral sclerosis in the Medicare programme and under commercial insurance. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(1-2):134-142. doi: 10.1080/21678421.2017.1363242
19. Nuedexta. Prescribing information. Avanir Pharmaceuticals, Inc; 2019. Accessed July 16, 2020. nuedexta.com/sites/default/files/Prescribing_Information.pdf
20. Bello-Haas VD. Physical therapy for individuals with amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis. 2018;8:45-54. doi: 10.2147/DNND.S146949
21. Rilutek. Prescribing information. Covis Pharmaceuticals; 2016. Accessed July 16, 2020. accessdata.fda.gov/drugsatfda_docs/label/2016/020599s017lbl.pdf
22. Tiglutik. Prescribing information. ITF Pharma; 2020. Accessed July 16, 2020. tiglutik.com/wp-content/uploads/2020/07/TIGLUTIK_PI_with_PEG.pdf
23. Exservan. Prescribing information. Aquestive Therapeutics; 2020. Accessed July 16, 2020.
www.accessdata.fda.gov/drugsatfda_docs/label/2020/212640s001lbl.pdf
24. Radicava. Prescribing information. Mitsubishi Tanabe Pharma Corporation; 2018. Accessed July 16, 2020. radicava.com/assets/dist/pdfs/radicava-prescribing-information.pdf
25. Aquestive Therapeutics receives FDA approval for Exservan (riluzole) oral film. News release. Aquestive Therapeutics, Inc; November 25, 2020. Accessed June 2, 2020. aquestive.com/aquestive-therapeutics-receives-fda-approval-for-exservan-riluzole-oral-film/
26. Ward AL, Sanjak M, Duffy K, et al. Power wheelchair prescription, utilization, satisfaction, and cost for patients with amyotrophic lateral sclerosis: preliminary data for evidence-based guidelines. Arch Phys Med Rehabil. 2010;91(2):268-272. doi: 10.1016/j.apmr.2009.10.023
27. Jones AR, Jivraj N, Balendra R, et al. Health utility decreases with increasing clinical stage in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(3-4):285-291. doi: 10.3109/21678421.2013.872149
28. Gruis KL, Chernew ME, Brown DL. The cost-effectiveness of early noninvasive ventilation for ALS patients. BMC Health Serv Res. 2005;5:58. doi: 10.1186/1472-6963-5-58
29. Ng L, Khan F, Young CA, Galea M. Symptomatic treatments for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2017;1(1):CD011776. doi: 10.1002/14651858.CD011776.pub2
30. Bromberg MB, Brownell AA, Forshew DA, Swenson M. A timeline for predicting durable medical equipment needs and interventions for amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler. 2010;11(1-2):110-115. doi: 10.3109/17482960902835970
31. Hwang CS, Weng HH, Wang LF, Tsai CH, Chang HT. An eye-tracking assistive device improves the quality of life for ALS patients and reduces the caregivers’ burden. J Mot Behav. 2014;46(4):233-238. doi: 10.1080/00222895.2014.891970
32. Roche JC, Rojas-Garcia R, Scott KM, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135(Pt 3):847-852. doi: 10.1093/brain/awr351
33. Fang T, Al Khleifat A, Stahl DR, et al. Comparison of the King’s and MiToS staging systems for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3-4):227-232. doi: 10.1080/21678421.2016.1265565
34. Millard J, Raza S, Boller E, Bauer E, Sullivan J, Apple S. Real-world healthcare utilization, costs, and progression milestone patterns among FFS Medicare patients newly diagnosed with amyotrophic lateral sclerosis (ALS). Poster presented at: ISPOR 2020; May 18-20, 2020. Accessed July 16, 2020. ispor.org/heor-resources/presentations-database/presentation/intl2020-3182/101102
35. Qutub K, Lacomis D, Albert SM, Feingold E. Life factors affecting depression and burden in amyotrophic lateral sclerosis caregivers. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(3-4):292-297. doi: 10.3109/21678421.2014.886699
36. Wang LH, Elliott MA, Jung Henson L, et al. Death with dignity in Washington patients with amyotrophic lateral sclerosis. Neurology. 2016;87(20):2117-2122. doi: 10.1212/WNL.0000000000003335
37. Frequently asked questions. Death with Dignity. Accessed April 28, 2020. deathwithdignity.org/faqs
38. Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler. 2010;11(1-2):178-180. doi: 10.3109/17482960903093710
39. A professional’s guide to assisting families with obtaining government benefits. ALS Association. Published April 27, 2020. Accessed April 28, 2020. alsa.org/als-care/resources/publications-videos/factsheets/professionals-guide-govt-benefits.html
40. Centers for Medicare and Medicaid Services. Chapter 6 – Part D Drugs and Formulary Requirements. In: Medicare Prescription Drug Benefit Manual. Centers for Medicare & Medicaid Services; January 15, 2016. Accessed April 28, 2020. www.cms.gov/Medicare/Prescription-Drug-Coverage/PrescriptionDrugCovContra/Downloads/Part-D-Benefits-Manual-Chapter-6.pdf
41. Facts about amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease). Muscular Dystrophy Association. Published May 2014. Accessed July 13, 2020. mda.org/sites/default/files/Facts_ALS_P-193_0.pdf
42. More information on certified centers and clinics. ALS Association. Accessed June 28, 2020. als.org/local-support/certified-centers-clinics/more-information-certified-centers-clinics
43. Ng L, Khan F, Mathers S. Multidisciplinary care for adults with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst Rev. 2009;(4):CD007425. doi: 10.1002/14651858.CD007425.pub2
44. Galvin M, Ryan P, Maguire S, et al. The path to specialist multidisciplinary care in amyotrophic lateral sclerosis: a population-based study of consultations, interventions and costs. PLoS One. 2017;12(6):e0179796. doi: 10.1371/journal.pone.0179796
45. Boylan K, Levine T, Lomen-Hoerth C, et al; ALS Center Cost Evaluation W/Standards & Satisfaction (Access) Consortium. Prospective study of cost of care at multidisciplinary ALS centers adhering to American Academy of Neurology (AAN) ALS practice parameters. Amyotroph Lateral Scler Frontotemporal Degener. 2015;17(1-2):119-127. doi: 10.3109/21678421.2015.1091478
46. Division of Neurology Products, Center for Drug Evaluation and Research. Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry. US Food and Drug Administration; September 2019. Accessed July 14, 2020. www.fda.gov/media/130964/download
47. Mazzini L, Ferrari D, Andjus PR, et al; BIONECA COST ACTION WG Neurology. Advances in
stem cell therapy for amyotrophic lateral sclerosis. Expert Opin Biol Ther. 2018;18(8):865-881.
doi: 10.1080/14712598.2018.1503248
48. Garrison LP, Jackson T, Paul D, Kenston M. Value-based pricing for emerging gene therapies: the economic case for a higher cost-effectiveness threshold. J Manag Care Spec Pharm. 2019;25(7):793-799. doi: 10.18553/jmcp.2019.18378
49. Cappella M, Ciotti C, Cohen-Tannoudji M, Biferi MG. Gene therapy for ALS-a perspective. Int J Mol Sci. 2019;20(18):4388. doi: 10.3390/ijms20184388
50. Institute for Clinical and Economic Review (ICER). Spinraza and Zolgensma for Spinal Muscular Atrophy: Effectiveness and Value. ICER; April 3, 2019. Accessed June 2, 2020. icer-review.org/wp-content/uploads/2018/07/ICER_SMA_Final_Evidence_Report_040319.pdf
51. Edaravone. Canadian Agency for Drugs and Technologies in Health (CADTH). Updated April 24, 2019. Accessed April 28, 2020. cadth.ca/edaravone
52. Shafrin J. Quality measurement in oncology: is the race worth running? Journal of Clinical Pathways blog. Published December 11, 2019. Accessed June 2, 2020. journalofclinicalpathways.com/commentary/quality-measurement-oncology-race-worth-running
53. Welcome to MIT NEWDIGS. New Drug Development ParadIGmS Initiative (NEWDIGS), Massachusetts Institute of Technology (MIT) Center for Biomedical Innovation. Accessed April 28, 2020. newdigs.mit.edu
54. MIT NEWDIGS FoCUS Project. Impact of patient mobility on annuity/performance-based contracting. MIT Center Biomedical Innovation. Published June 15, 2018. Accessed April 28, 2020. newdigs.mit.edu/sites/default/files/FoCUS_Research_Brief_2018F206v022.pdf
Click here to download PDF
