
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Real-World Data on the Course of Idiopathic Pulmonary Fibrosis

Abstract
Idiopathic pulmonary fibrosis (IPF) is characterized by a progressive decline in lung function, worsening quality of life, and high mortality. However, the rate and pattern of progression of IPF are variable. Real-world studies, which include a broader population of patients than clinical trials and collect data over longer periods, have provided important information on the clinical course of IPF and further insights into the efficacy and safety of antifibrotic therapies. They also highlight the worsening of patients’ quality of life as lung function is lost, the high frequency of hospitalizations, and the impact of acute exacerbations on mortality in patients with IPF. Data from patient registries and analyses of claims data suggest that antifibrotic therapy is more likely to be used in patients who have worse lung function and that its use is associated with an improvement in life expectancy. The safety profile of antifibrotic therapies in real-world populations is consistent with that observed in clinical trials. Further real-world studies are needed to improve understanding of the course and impact of IPF in specific groups of patients and how the care provided to these patients might be improved.
Am J Manag Care. 2024;30:S107-S113. https://doi.org/10.37765/ajmc.2024.89632
For author information and disclosures, see end of text.



Introduction
Idiopathic pulmonary fibrosis (IPF) is a type of interstitial lung disease (ILD) that is progressive over time.1 However, the rate at which IPF progresses is variable and its trajectory can be slowed by the use of antifibrotic drugs.2,3 Real-world studies have informed our knowledge of the course of IPF. In this article, we discuss the data from those studies, with a focus on recent evidence.
Delays in Diagnosis of IPF
A diagnosis of IPF is often not made for some time after the onset of symptoms. A survey of US physicians conducted from 2018 to 2019 suggested that the typical delay from symptom onset to diagnosis of IPF was 1.7 years.4 Among 347 patients enrolled in a US registry between 2014 and 2019, approximately 50% were diagnosed more than a year after symptom onset.5 A diagnosis of IPF is often delayed due to an incorrect diagnosis having been made. In a retrospective study of Medicare claims data from 44,891 US patients between 2014 and 2019, a diagnosis of IPF was preceded by other respiratory diagnoses in almost all cases, and the mean time from an initial respiratory diagnosis to a diagnosis of IPF was 2.7 years.6
Delays in diagnosis may result in lung function being significantly impaired by the time IPF is diagnosed. Among 253 incident cases of IPF in the Canadian Registry for Pulmonary Fibrosis (CAREPF), the mean forced vital capacity (FVC) was 73% predicted and the mean diffusing capacity for carbon monoxide (DLCO) was 50% predicted.7 Similarly, among 498 patients who received a new diagnosis of IPF at the enrolling center for the US IPF-PRO Registry, the median FVC at enrollment was 71% predicted and the median DLCO was 42% predicted.5 There is some evidence that a delayed diagnosis of IPF is associated with worse outcomes. A study of 264 patients diagnosed with IPF at 2 specialist ILD centers in Denmark from April 2016 to June 2021 found that patients who were not diagnosed with IPF for more than a year after their first IPF-related symptom had a higher rate of hospital admissions (0.7 vs 0.6 admissions per person-year) and worse progression-free survival (HR, 1.70; 95% CI, 1.18-2.46; P = .004) than patients diagnosed less than a year after their first IPF-related symptom, over a follow-up period of up to 5 years.8 However, in an analysis of data from 498 patients in the US IPF-PRO Registry, there was no significant difference in time to death or lung transplant between patients who were diagnosed more than versus less than 1 year after symptom onset.5
Progression of IPF
IPF is associated with a progressive decline in FVC, but FVC does not decline at the same rate in all patients and patients may experience periods of stability or even slight increases in FVC.9,10 A machine learning–based analysis of data from 415 patients with IPF in a multicenter UK study (with IPF diagnosed ≤ 6 months prior ) or at the University of Chicago (with IPF diagnosed ≥ 3 months prior), identified 4 clusters of patients with different trajectories of FVC over 3 years. In 34% of the cohort, there was a linear decline in FVC, in 24% there was an initial improvement followed by a decline, in 27% there was an initial decline then stabilization, while stability was evident in 15% of the cohort.10 Joint modeling of data from over 900 patients in the IPF-PRO Registry that were collected over a median follow-up period of almost 3 years suggested that mean declines in the FVC percentage predicted and DLCO percentage predicted were 2.8% per year and 2.9% per year, respectively.11 A retrospective study of data from 249 patients on antifibrotic therapy (nintedanib or pirfenidone) referred to 3 ILD centers in Italy between 2011 and 2017 found an FVC decline over the first year of approximately 100 mL, with no significant difference between patients with a definite usual interstitial pneumonia (UIP) pattern on high-resolution CT (HRCT) or biopsy and those with a possible UIP pattern on HRCT.12
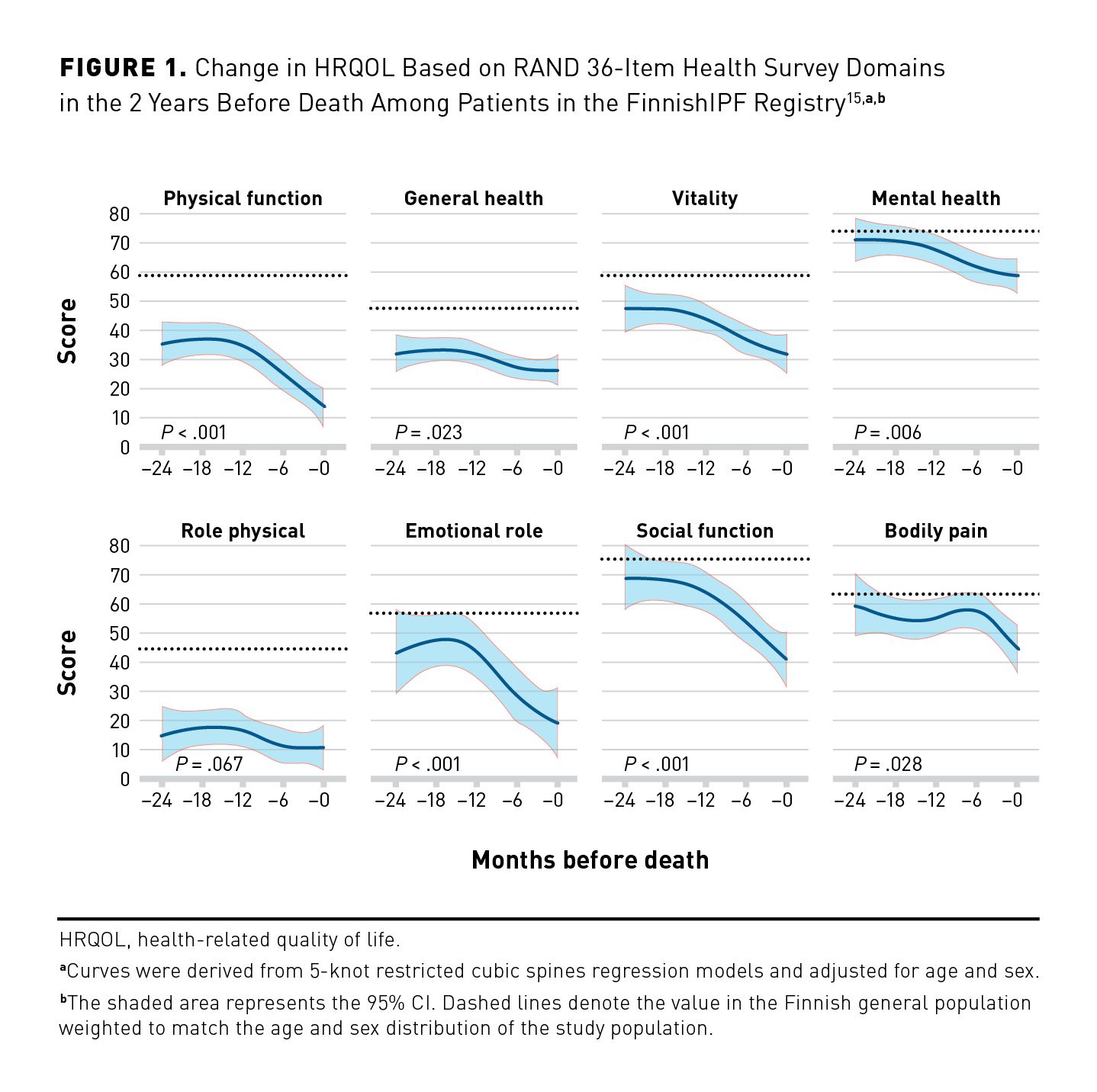
As IPF progresses, patients experience deterioration in health-related quality of life (HRQOL) due to worsening of symptoms and deterioration in physical function. At enrollment into the IPF-PRO Registry, a lower FVC percentage predicted was associated with worse HRQOL based on the St George’s Respiratory Questionnaire (SGRQ) score.13 Among 424 patients with IPF in the German INSIGHTS-IPF registry, HRQOL worsened over 3 years based on several patient-reported outcomes, including the SGRQ total score (mean change, 1.47; 95% CI, 1.17- 1.76; P < .001).14 Data from 92 patients in the Finnish IPF Registry showed significant increases in the severity of impairment in several domains of physical and emotional health during the last 24 months of life (Figure 1).15


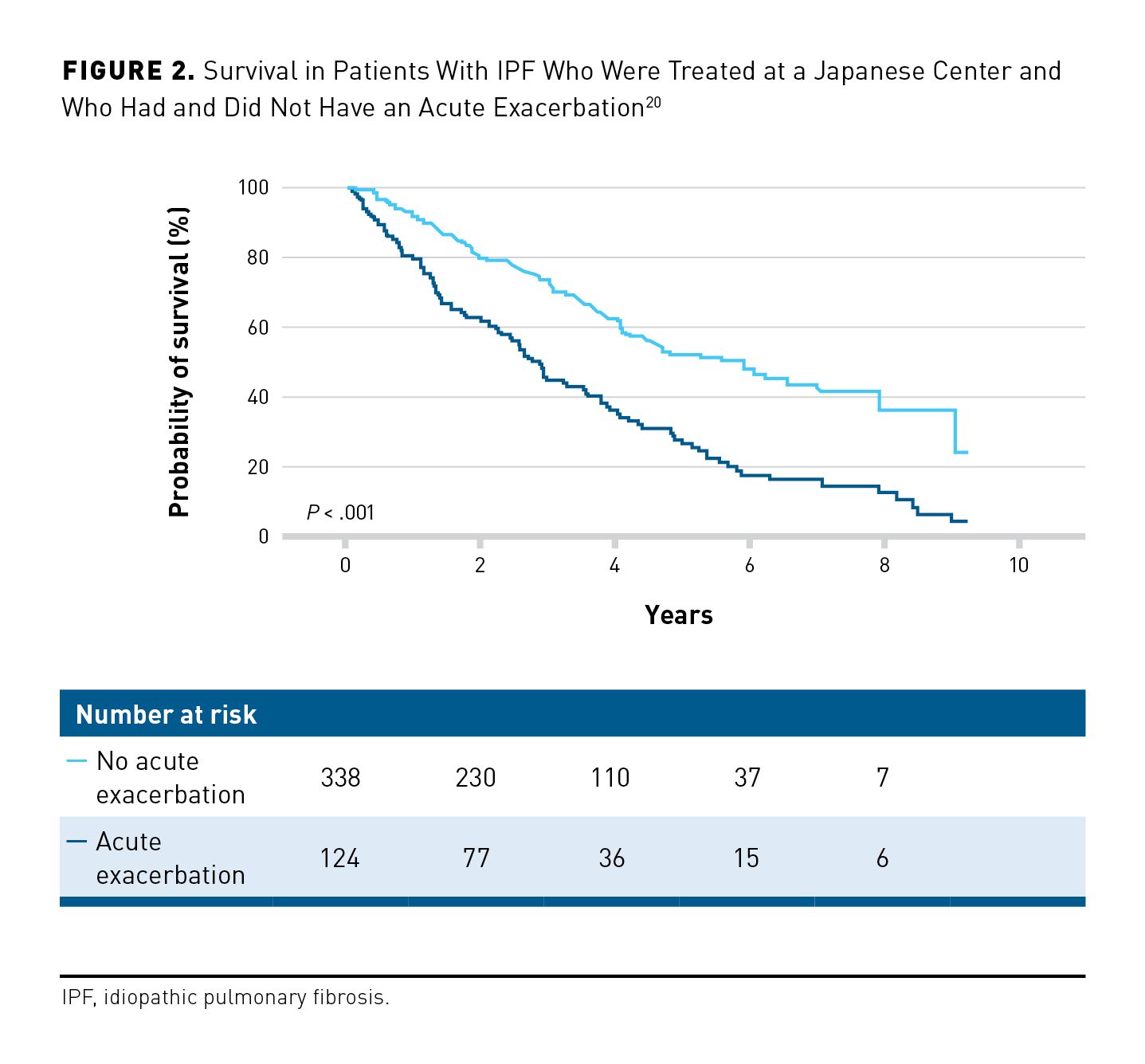
A minority of patients with IPF experience acute deteriorations in lung function, accompanied by worsening of radiologic abnormalities, which are known as acute exacerbations.16 In a retrospective analysis of data from 195 Japanese patients, the cumulative incidence of acute exacerbations in the 1, 2, and 3 years following initiation of antifibrotic therapy was 9%, 22%, and 25%, respectively.17 Similarly, in an analysis of claims data from 9961 newly diagnosed Japanese patients with IPF, the annual incidence of acute exacerbations was approximately 10%.18 In a prospective analysis of data from 676 Italian patients with newly diagnosed IPF, of whom 40% received antifibrotic therapy, 37% experienced at least 1 acute exacerbation over 5 years.19 Acute exacerbations are associated with high short-term mortality.20,21 In an analysis of data from 462 Japanese patients evaluated at a Japanese referral center, the median survival after evaluation at the center was 2.9 years in patients who had an acute exacerbation versus 5.8 years for those who did not (P < .001) (Figure 2); 90-day mortality following an acute exacerbation was 47%.20


Utility of Antifibrotic Therapy
Two antifibrotic therapies were approved in the US for the treatment of IPF in 2014: nintedanib and pirfenidone. In an analysis of claims data from 10,996 US patients with IPF collected between October 2014 and July 2019, approximately 26% had started antifibrotic therapy.22 More recent data from a patient registry in Canada suggest that 54% of patients with IPF are receiving antifibrotic therapy.23 Data from a patient registry in the US suggest that 70% of patients with IPF are receiving antifibrotic therapy and that its use is more frequent in patients who are younger (OR, 0.87; 95% CI, 0.79-0.97, per 5-year higher), have worse FVC percentage predicted (OR, 0.91; 95% CI, 0.84-1.00, per 10% higher), or use supplemental oxygen with activity (OR, 1.55; 95% CI, 1.11-2.16).24 However, most of the patients in these registries were receiving their care at specialized centers, and the use of antifibrotic therapy in the community is likely to be lower. In a US cohort of 14,792 veterans with IPF, patients were less likely to receive antifibrotic therapy if they lived in a rural area (OR, 0.88; 95% CI, 0.80-0.97; P = .012) or received their care outside of the Veterans Affairs Healthcare System (OR, 0.15; 95% CI, 0.10-0.22; P < .001).25
Real-world studies provide data on the use of antifibrotic therapies in broader patient populations than those enrolled in clinical trials. These include patients who are elderly, have severe lung function impairment, have several comorbidities, and are taking medications for comorbid conditions. Data from 2 single-center Korean studies suggested that antifibrotic therapy reduced FVC decline both in patients with advanced lung function impairment (FVC < 50% predicted and/or DLCO < 35% predicted) and in those with more preserved lung function, with similar adverse event profiles.26 Among 1009 patients enrolled in the German INSIGHTS-IPF registry, the decline in FVC percentage predicted over the 24 months after initiation of antifibrotic therapy was similar in patients aged ≥75 versus those aged < 75 years (difference, –0.05; 95% CI, –0.20 to 0.09; P = .478).27 Data from multiple real-world studies suggest that the safety and tolerability profiles of antifibrotic therapies in clinical practice are similar to those observed in clinical trials.28-31
Real-world studies also shed light on the rate of discontinuation of antifibrotic therapy when prescribed in clinical practice. Among 703 patients with IPF enrolled in the US Pulmonary Fibrosis Foundation Patient Registry, 11% had discontinued antifibrotic therapy within the 12 months prior to enrollment.24 In the Finnish IPF registry, 22% of patients were no longer taking antifibrotic therapy 1 year after drug initiation.32 In an Italian study of 263 patients, 8% of subjects discontinued antifibrotic therapy due to adverse events over an average follow-up of almost 2 and a half years.30 Real-world studies have revealed factors associated with a higher risk of discontinuation of antifibrotic therapy. In a study of 104 patients in the UK, a number of factors were associated with discontinuation of antifibrotic therapy over 12 months including age 75 years or older (OR, 3.17; 95% CI, 1.16-8.68; P = .025), female sex (OR, 4.57; 95% CI, 1.40-14.88; P = .012), and BMI ≤25 kg/m2 (OR, 3.74; 95% CI, 1.22-11.53; P = .021).33 Pharmacovigilance data from 219 patients who received pirfenidone in Korea suggested that the rate of discontinuation was higher among those with FVC <50% predicted and/or DLCO <35% predicted at baseline than in those with less severe impairment in lung function (74% vs 50%; P = .006).34 In a study of 104 Japanese patients, poor overall health (performance status) at initiation of nintedanib was a risk factor for treatment discontinuation over 12 months (OR, 0.014; 95% CI, 0.002-0.167; P = .007).35
Hospitalizations in Patients With IPF
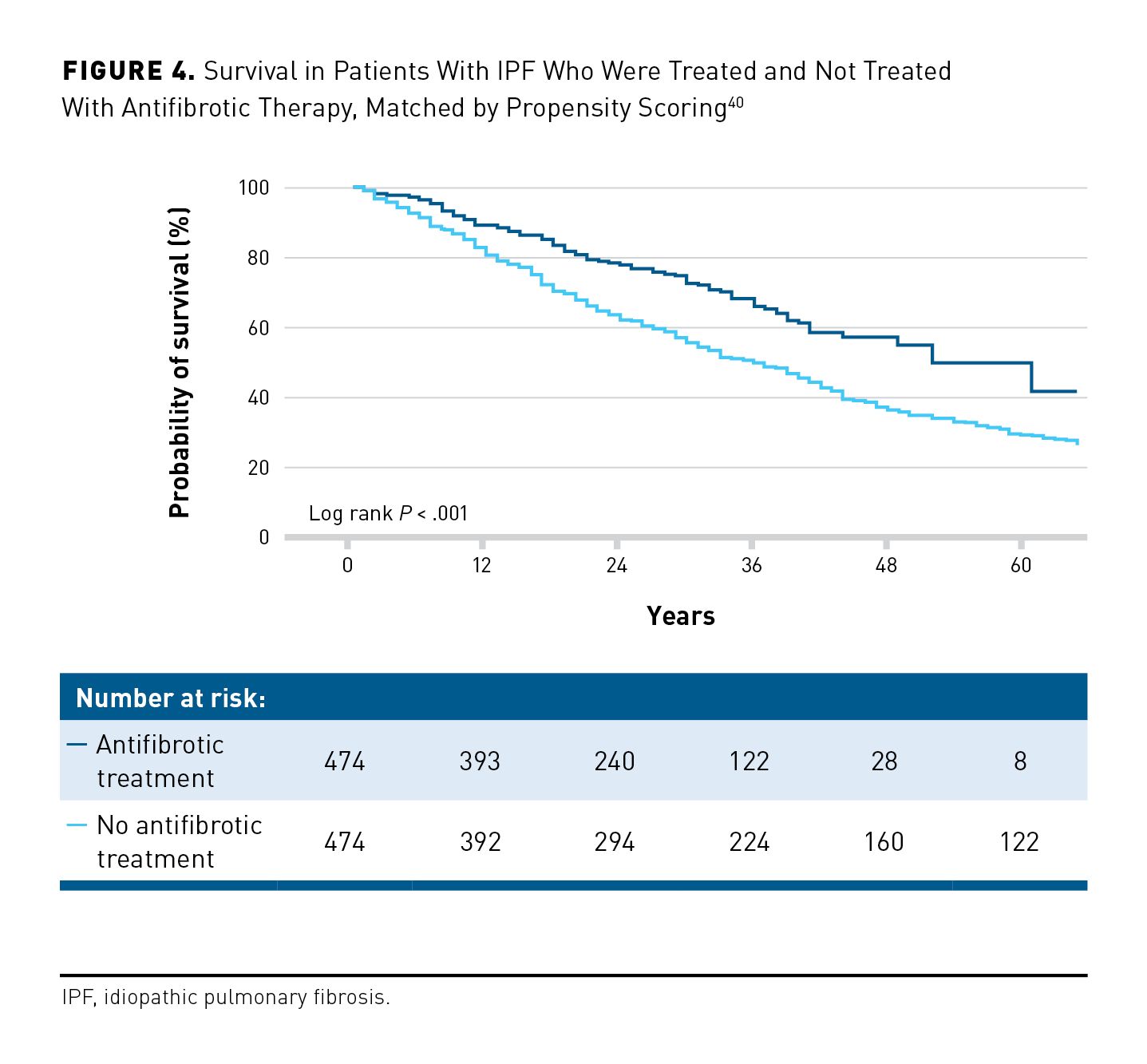
In patients with IPF, hospitalizations are frequent and have a strong association with mortality. Among 1002 patients enrolled in the US IPF-PRO Registry, 57% had at least 1 hospitalization (of any cause) over a median follow-up of 23.7 months.36 Risk factors associated with hospitalization during follow-up included lower DLCO percentage predicted, oxygen use at rest, and hospitalization in the year prior to enrollment. There was an 8-fold increase in the risk of mortality during hospitalization or within 90 days of discharge compared with the risk of mortality outside this period (P <.001).36 An analysis of administrative data from 93,680 US patients with IPF hospitalized between 2013 and 2019 found that the in-hospital mortality rate was 11%.37 Similarly, an analysis of claims data from 9667 US patients hospitalized between 2015 and 2018 found that 11% of patients died during the first hospitalization, while 29% of patients were readmitted to the hospital within 90 days of that first visit.38 In-hospital mortality rates in patients with IPF are particularly high in patients who receive mechanical ventilation.36,38,39 In the same dataset, mechanical ventilation was associated with a more than 6-fold increase in the risk of in-hospital mortality or lung transplant (OR, 6.41; 95% CI, 5.24-7.84).38 There is some evidence that the use of antifibrotic therapy might reduce the rate of hospitalization in patients with IPF. In a study of 948 patients in which treated and untreated patients were matched using propensity scores, the proportion of patients with respiratory-related hospitalization over 27 months was 22.2% in the treated group and 36.9% in the untreated group (P <.001).40 An analysis of claims data from 9282 US patients also suggested that antifibrotic therapy was associated with a lower risk of both all-cause (HR, 0.71; 95% CI, 0.67-0.76) and respiratory-related (HR, 0.70; 95% CI, 0.64-0.76) hospitalization.41
Survival in Patients With IPF
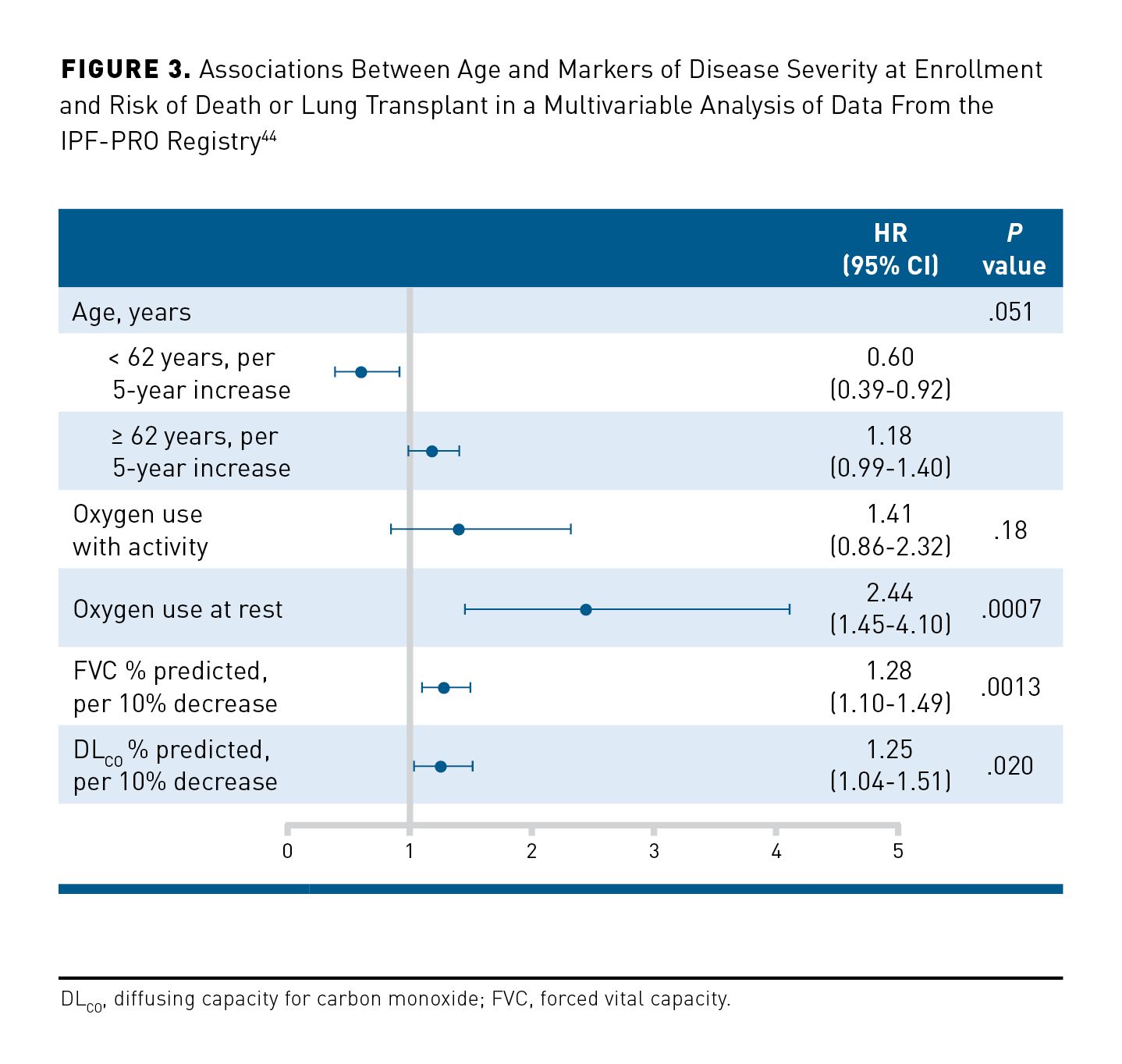
Recent real-world studies provide the best estimates of life expectancy following a diagnosis of IPF. In a Korean patient registry, the 3-year survival rate among 1345 patients diagnosed with IPF between 2012 and 2018 was 70%.42 In a Danish study, median survival among 260 patients diagnosed with IPF between 2011 and 2016 was 4.0 years, compared with 3.2 years among 121 patients diagnosed between 2003 and 2009.43 While observations vary across studies, male sex, a greater extent of fibrosis on CT, worse lung function, worse symptoms/HRQOL, and weight loss have been associated with increased short-term mortality.44-50 Among 662 patients in the US IPF-PRO Registry, oxygen use at rest (HR, 2.44; 95% CI, 1.45-4.10; P = .0007), lower FVC percentage predicted (HR, 1.28; 95% CI, 1.10-1.49; P = .0013, per 10% lower), and lower DLCO percentage predicted (HR, 1.25; 95% CI, 1.04-1.51; P = .020, per 10% lower) were significantly associated with an increased risk of death or lung transplant over a follow-up period of 30 months (Figure 3).44 Furthermore, patients with IPF commonly have comorbid conditions such as pulmonary hypertension and cardiovascular disease that place them at risk for increased mortality.51 Among 272 patients treated at a tertiary referral center in Germany, median survival was 66 months in patients without comorbidities, 48 months in patients with up to 3 comorbidities and 35 months for patients with 4 to 7 comorbidities.52 In a study of 3580 patients with IPF enrolled in the EMPIRE registry, pulmonary hypertension, stroke, respiratory infection, emphysema, lung cancer, and obesity were associated with an increased risk of mortality over a median follow-up of 13.8 months.53


Analyses of data from registries, other observational studies, and insurance claims have shown a reduced risk of mortality in patients with IPF who receive antifibrotic therapy.19,40-42,53-56 These studies should be interpreted with caution, as they are based on comparison of nonrandomized groups and, in most cases, are affected by immortal time bias and biases arising from the initiation or discontinuation of antifibrotic therapy during follow-up.57 To avoid such biases, an analysis of data from 499 patients in the US IPF-PRO Registry used a new-user design, comparing newly-treated patients with never-treated patients, and a causal inference methodology, which accounted for treatment initiations and discontinuations during follow-up. This analysis found a 47% reduction in the risk of death in the patients who received treatment (HR, 0.53; 95% CI, 0.28-1.03; P = .060).56 Similarly, in a study of 948 patients, in which propensity scores were used to match the treated and untreated patients, use of antifibrotic therapy was associated with a 41% reduction in the risk of mortality over 27 months (HR, 0.59; 95% CI, 0.48-0.72; P < .001) (Figure 4).40 An analysis of claims data from 48,638 patients with IPF, based on a time-varying Cox proportional hazards model, suggested that use of antifibrotic therapy reduced mortality by 31% compared with never receiving treatment (HR, 0.69; 95% CI, 0.66-0.72; P < 0.001).58 A study of claims data from 9282 patients aged at least 67 years suggested that antifibrotic therapy provided a similar reduction in the risk of mortality in this age group (HR, 0.62; 95% CI, 0.57-0.68).41


The growing real-world evidence suggesting that the use of antifibrotic therapy is associated with reduced mortality underscores the value of patient registries and other big data sources in clarifying the role of therapies post approval. This is especially important in IPF, where the approval of antifibrotic drugs was based on the surrogate end point of change in FVC,59,60 and change in FVC continues to be the end point used in most clinical trials. Real-world studies, as well as further analyses of data from clinical trials,61 are providing additional evidence to support the use of change in FVC as the end point to assess the efficacy of drugs to treat IPF.
Lung Transplant in Patients With IPF
The International Society for Heart and Lung Transplantation (ISHLT) guidelines recommend that any patient with histopathologic or radiographic evidence of UIP is appropriate for lung transplant referral, and that evaluation for lung transplant should include a thorough assessment of the severity of lung disease, frailty, the presence and severity of comorbidities, and psychosocial circumstances.62 In 2023, 986 patients with IPF received a lung transplant in the US.63 However, patients with IPF still comprise about 20% of patients on the wait list.63 A multivariable analysis of data from 475 patients with IPF in the US Pulmonary Fibrosis Foundation (PFF) Registry found that younger age, lower FVC percentage predicted, and use of supplemental oxygen at rest were predictors of being placed on a list for lung transplant.64 Socioeconomic and geographic factors may also play a role.65,66 An analysis of data from the US IPF-PRO Registry showed that, irrespective of eligibility factors, patients who lived in a zip code with higher median household income or who were enrolled at a center with a lung transplant program had a higher likelihood of receiving a lung transplant.66
The number of lung transplants performed in patients with IPF is increasing. In the US, the number of lung transplants performed in patients with IPF increased from 231 in 2003 to 984 in 2023.63 This likely reflects the increasing upper age limit for lung transplants at most transplant centers. In addition, the greater availability of extracorporeal membrane oxygenation as a bridge to transplant has enabled more end-stage patients to survive to transplant. The number of transplantable organs is also increasing, with novel technologies to preserve and resuscitate lungs, such as ex-vivo lung perfusion. Furthermore, there have been modifications to the lung allocation score, updated in March 2023 with the implementation of the continuous allocation score,67 which aim to reduce deaths on the waiting list, increase transplants for the sickest patients, and reduce geographic variability in access to transplants.
There has been concern that the use of antifibrotic therapy close to lung transplant may increase the risk of bleeding or delay wound healing. However, observational studies suggest that the use of antifibrotic therapy before transplant surgery does not have a negative impact on the incidence of complications during and following the surgery.68-71 For example, a study of data from 297 US patients taking nintedanib (n = 107) or pirfenidone (n = 190) found that the use of antifibrotic therapy until ≤5 drug half-lives before lung transplant surgery did not affect the incidence of surgical wound dehiscence or survival to discharge.71
Conclusions
Real-world studies provide important information on the course, impact, and treatment of IPF in the varied population of patients managed in clinical practice. These studies illustrate the heavy burden that IPF places on patients and health care resources, the variability in the course of the disease, and the utility of antifibrotic therapies in improving outcomes. Clinical trials are, by their nature, “discriminatory” in that they exclude patients who do not meet strict inclusion and exclusion criteria. Real-world studies serve as the backfill to evaluate how these drugs perform in a more general population. These studies are important not only to provide further support for the utility of these drugs, but also to identify unmet needs, systemic inequities, and areas for future research. Future real-world studies in patients with IPF are strongly encouraged since they may help improve the provision of care for all patients with this devastating disease.
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors. Editorial support was provided by Julie Fleming and Wendy Morris of Fleishman-Hillard, London, United Kingdom, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Author Affiliations: Department of Medicine, University Colorado Denver Anschuntz Medical Campus (JSL), Aurora, CO; Inova Advances Lung Disease and Transplant Program, Inova Fairfax Hospital (SDN), Falls Church, VA.
Source of Funding: This article is part of a supplement that was supported by Boehringer Ingelheim Pharmaceuticals, Inc. The authors did not receive any payment for their work on the articles. Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Author Disclosures: Dr Lee reports participating in consultancies or paid advisory boards for Boehringer Ingelheim. Dr Nathan reports participating in consultancies or paid advisory boards for Boehringer Ingelheim and United Therapeutics. He also reports receiving lecture fees for speaking at the invitation of Boehringer Ingelheim and United Therapeutics.
Authorship Information: Concept and design (JSL, SDN); acquisition of data (SDN); analysis and interpretation of data (JSL, SDN); drafting of the manuscript (SDN); critical revision of the manuscript for important intellectual content (JSL, SDN).
Address Correspondence to: Steven D. Nathan, MD. Inova Fairfax Hospital, 3300 Gallows Rd, Falls Church, VA, 22042. Email: steven.nathan@inova.org
REFERENCES
1. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47. doi:10.1164/rccm.202202-0399ST
2. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082. doi: 10.1056/NEJMoa1402584
3. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083-2092. doi:10.1056/NEJMoa1402582
4. Lancaster L, Bonella F, Inoue Y, et al. Idiopathic pulmonary fibrosis: physician and patient perspectives on the pathway to care from symptom recognition to diagnosis and disease burden. Respirology. 2022;27(1):66-75. doi:10.1111/resp.14154
5. Snyder LD, Mosher C, Holtze CH, et al. Time to diagnosis of idiopathic pulmonary fibrosis in the IPFPRO Registry. BMJ Open Respir Res. 2020;7(1):e000567. doi:10.1136/bmjresp-2020-000567
6. Herberts MB, Teague TT, Thao V, et al. Idiopathic pulmonary fibrosis in the United States: time to diagnosis and treatment. BMC Pulm Med. 2023;23(1):281. doi:10.1186/s12890-023-02565-7
7. Fisher JH, Kolb M, Algamdi M, et al. Baseline characteristics and comorbidities in the CAnadian REgistry for Pulmonary Fibrosis. BMC Pulm Med. 2019;19(1):223. doi:10.1186/s12890-019-0986-4
8. Hoyer N, Prior TS, Bendstrup E, et al. Diagnostic delay in IPF impacts progression-free survival, quality of life and hospitalisation rates. BMJ Open Respir Res. 2022;9(1):e001276. doi:10.1136/bmjresp-2022-001276
9. Flaherty KR, Kolb M, Vancheri C, et al. Stability or improvement in forced vital capacity with nintedanib in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2018;52(2):1702593. doi:10.1183/13993003.02593-2017
10. Fainberg HP, Oldham JM, Molyneaux PL, et al. Forced vital capacity trajectories in patients with idiopathic pulmonary fibrosis: a secondary analysis of a multicentre, prospective, observational cohort. Lancet Digit Health. 2022;4(12):e862–872. doi:10.1016/S2589-7500(22)00173-X
11. Neely ML, Hellkamp AS, Bender S, et al. Lung function trajectories in patients with idiopathic pulmonary fibrosis. Respir Res. 2023;24(1):209. doi:10.1186/s12931-023-02503-5
12. Sgalla G, Lo Greco E, Calvello M, et al. Disease progression across the spectrum of idiopathic pulmonary fibrosis: a multicentre study. Respirology. 2020;25(11):1144-1151. doi:10.1111/resp.13805
13. O’Brien EC, Hellkamp AS, Neely ML, et al. Disease severity and quality of life in patients with idiopathic pulmonary fibrosis: a cross-sectional analysis of the IPF-PRO Registry. Chest. 2020;157(5):1188-1198. doi:10.1016/j.chest.2019.11.042
14. Kreuter M, Swigris J, Pittrow D, et al. The clinical course of idiopathic pulmonary fibrosis and its association to quality of life over time: longitudinal data from the INSIGHTS-IPF registry. Respir Res. 2019;20(1):59. doi:10.1186/s12931-019-1020-3
15. Rajala K, Lehto JT, Sutinen E, et al. Marked deterioration in the quality of life of patients with idiopathic pulmonary fibrosis during the last two years of life. BMC Pulm Med. 2018:18:172. doi:10.1186/s12890-018-0738-x
16. Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265-275. doi:10.1164/rccm.201604-0801CI
17. Isshiki T, Sakamoto S, Yamasaki A, et al. Incidence of acute exacerbation of idiopathic pulmonary fibrosis in patients receiving antifibrotic agents: real-world experience. Respir Med. 2021;187:106551. doi:10.1016/j.rmed.2021.106551
18. Homma S, Suda T, Hongo Y, et al. Incidence and changes in treatment of acute exacerbation of idiopathic pulmonary fibrosis in Japan: a claims-based retrospective study. Respir Investig. 2022;60(6):798-805. doi:10.1016/j.resinv.2022.07.004
19. Iommi M, Faragalli A, Bonifazi M, et al. Prognosis and survival in idiopathic pulmonary fibrosis in the era of antifibrotic therapy in Italy: evidence from a longitudinal population study based on healthcare utilization databases. Int J Environ Res Public Health. 2022;19(24):16689. doi:10.3390/ijerph192416689
20. Suzuki A, Kondoh Y, Brown KK, et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology. 2020;25(5):525-534. doi:10.1111/resp.13682
21. Yoo JW, Kim J, Song JW. Impact of the revised definition on incidence and outcomes of acute exacerbation of idiopathic pulmonary fibrosis. Sci Rep. 2022;12(1):8817. doi:10.1038/s41598-022-12693-5
22. Dempsey TM, Payne S, Sangaralingham L, et al. Adoption of the antifibrotic medications pirfenidone and nintedanib for patients with idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2021;18:1121-1128. doi:10.1513/AnnalsATS.202007-901OC
23. Assayag D, Garlick K, Johannson KA, et al. Treatment initiation in patients with interstitial lung disease in Canada. Ann Am Thorac Soc. 2021;18(10):1661-1668. doi:10.1513/AnnalsATS.202009-1122OC
24. Salisbury ML, Conoscenti CS, Culver DA, et al. Antifibrotic drug use in patients with idiopathic pulmonary fibrosis. Data from the IPF-PRO Registry. Ann Am Thorac Soc. 2020;17(11):1413-1423. doi:10.1513/AnnalsATS.201912-880OC
25. Kaul B, Lee JS, Petersen LA, et al. Disparities in antifibrotic medication utilization among veterans with idiopathic pulmonary fibrosis. Chest. 2023;164(2):441-449. doi:10.1016/j.chest.2023.02.027
26. Yoon HY, Kim DS, Song JW. Efficacy and safety of pirfenidone in advanced idiopathic pulmonary fibrosis. Respiration. 2019;97(3):242-251. doi:10.1159/000492937
27. Leuschner G, Klotsche J, Kreuter M, et al. Idiopathic pulmonary fibrosis in elderly patients: analysis of the INSIGHTS-IPF observational study. Front Med (Lausanne). 2020;7:601279. doi:10.3389/fmed.2020.601279
28. Tzouvelekis A, Karampitsakos T, Ntolios P, et al. Longitudinal “real-world” outcomes of pirfenidone in idiopathic pulmonary fibrosis in Greece. Front Med (Lausanne). 2017;4:213. doi:10.3389/fmed.2017.00213
29. Vancheri C, Sebastiani A, Tomassetti S, et al. Pirfenidone in real life: a retrospective observational multicentre study in Italian patients with idiopathic pulmonary fibrosis. Respir Med. 2019;156:78-84. doi:10.1016/j.rmed.2019.08.006
30. Cameli P, Refini RM, Bergantini L, et al. Long-term follow-up of patients with idiopathic pulmonary fibrosis treated with pirfenidone or nintedanib: a real-life comparison study. Front Mol Biosci. 2020;7:581828. doi:10.3389/fmolb.2020.581828
31. Podolanczuk AJ, Cottin V. A narrative review of real-world data on the safety of nintedanib in patients with idiopathic pulmonary fibrosis. Adv Ther. 2023;40(5):2038-2050. doi:10.1007/s12325-023-02454-9
32. Kaunisto J, Salomaa ER, Koivisto M, et al. Overall drug treatment of idiopathic pulmonary fibrosis patients from national registries - a real-world study from Finland. BMC Pulm Med. 2023;23(1):364. doi:10.1186/s12890-023-02630-1
33. Wright WA, Crowley LE, Parekh D, et al. Real-world retrospective observational study exploring the effectiveness and safety of antifibrotics in idiopathic pulmonary fibrosis. BMJ Open Respir Res. 2021;8(1):e000782. doi:10.1136/bmjresp-2020-000782
34. Chung MP, Park MS, Oh IJ, et al. Safety and efficacy of pirfenidone in advanced idiopathic pulmonary fibrosis: a nationwide post-marketing surveillance study in Korean patients. Adv Ther. 2020;37(5):2303-2316. doi:10.1007/s12325-020-01328-8
35. Kato M, Sasaki S, Tateyama M, et al. Clinical significance of continuable treatment with nintedanib over 12 months for idiopathic pulmonary fibrosis in a real-world setting. Drug Des Devel Ther. 2021;15:223-230. doi:10.2147/DDDT.S284819
36. Kim HJ, Snyder LD, Adegunsoye A, et al. Hospitalizations in patients with idiopathic pulmonary fibrosis. Respir Res. 2021;22(1):257. doi:10.1186/s12931-021-01851-4
37. Alqalyoobi S, Fernández Pérez ER, Oldham JM. In-hospital mortality trends among patients with idiopathic pulmonary fibrosis in the United States between 2013-2017: a comparison of academic and non-academic programs. BMC Pulm Med. 2020;20(1):289. doi:10.1186/s12890-020-01328-y
38. Durheim MT, Judy J, Bender S, et al. A retrospective study of in-hospital mortality in patients with idiopathic pulmonary fibrosis between 2015 and 2018. Medicine (Baltimore). 2020;99(47):e23143. doi:10.1097/MD.0000000000023143
39. Mooney JJ, Raimundo K, Chang E, et al. Mechanical ventilation in idiopathic pulmonary fibrosis: a nationwide analysis of ventilator use, outcomes, and resource burden. BMC Pulm Med. 2017;17(1):84. doi:10.1186/s12890-017-0426-2
40. Kang J, Han M, Song JW. Antifibrotic treatment improves clinical outcomes in patients with idiopathic pulmonary fibrosis: a propensity score matching analysis. Sci Rep. 2020;10(1):15620. doi:10.1038/s41598-020-72607-1
41. Mooney J, Reddy SR, Chang E, et al. Antifibrotic therapies reduce mortality and hospitalization among Medicare beneficiaries with idiopathic pulmonary fibrosis. J Manag Care Spec Pharm. 2021;27(12):1724-1733. doi:10.18553/jmcp.2021.27.12.1724
42. Moon SW, Kim SY, Chung MP, et al. Longitudinal changes in clinical features, management, and outcomes of idiopathic pulmonary fibrosis. A nationwide cohort study. Ann Am Thorac Soc. 2021;18(5):780-787. doi:10.1513/AnnalsATS.202005-451OC
43. Hyldgaard C, Møller J, Bendstrup E. Changes in management of idiopathic pulmonary fibrosis: impact on disease severity and mortality. Eur Clin Respir J. 2020;7(1):1807682. doi:10.1080/20018525.2020
44. Snyder L, Neely ML, Hellkamp AS, et al. Predictors of death or lung transplant in patients with IPF: data from the IPF-PRO Registry. Respir Res. 2019;20(1):105. doi:10.1186/s12931-019-1043-9
45. Kaunisto J, Salomaa ER, Hodgson U, et al. Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res. 2019;5(3):00170-2018. doi:10.1183/23120541.00170-2018
46. Case AH, Hellkamp AS, Neely ML, et al. Associations between patient-reported outcomes and death or lung transplant in IPF: data from the IPF-PRO Registry. Ann Am Thorac Soc. 2020;17(6):699-705. doi:10.1513/AnnalsATS.201906-437OC
47. Zaman T, Moua T, Vittinghoff E, et al. Differences in clinical characteristics and outcomes between men and women with idiopathic pulmonary fibrosis: a multicenter retrospective cohort study. Chest. 2020;158(1):245-251. doi:10.1016/j.chest.2020.02.009
48. Gao J, Kalafatis D, Carlson L, et al. Baseline characteristics and survival of patients of idiopathic pulmonary fibrosis: a longitudinal analysis of the Swedish IPF Registry. Respir Res. 2021;22(1):40. doi:10.1186/s12931-021-01634-x
49. Humphries SM, Mackintosh JA, Jo HE, et al. Quantitative computed tomography predicts outcomes in idiopathic pulmonary fibrosis. Respirology. 2022;27(12):1045-1053. doi:10.1111/resp.14333
50. Lee JH, Park HJ, Kim S, et al. Epidemiology and comorbidities in idiopathic pulmonary fibrosis: a nationwide cohort study. BMC Pulm Med. 2023;23(1):54. doi:10.1186/s12890-023-02340-8
51. King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med. 2017;5(1):72-84. doi:10.1016/S2213-2600(16)30222-3
52. Kreuter M, Ehlers-Tenenbaum S, Palmowski K, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11(3):e0151425. doi:10.1371/journal.pone.0151425
53. Jovanovic DM, Šterclová M, Mogulkoc N, et al. Comorbidity burden and survival in patients with idiopathic pulmonary fibrosis: the EMPIRE registry study. Respir Res. 2022;23(1):135. doi:10.1186/s12931-022-02033-6
54. Behr J, Prasse A, Wirtz H, et al. Survival and course of lung function in the presence or absence of antifibrotic treatment in patients with idiopathic pulmonary fibrosis: long-term results of the INSIGHTS-IPF registry. Eur Respir J. 2020;56(2):1902279. doi:10.1183/13993003.02279-2019
55. Pastre J, Barnett S, Ksovreli I, et al. Idiopathic pulmonary fibrosis patients with severe physiologic impairment: characteristics and outcomes. Respir Res. 2021;22(1):5. doi:10.1186/s12931-020-01600-z
56. de Andrade JA, Neely ML, Hellkamp AS, et al. Effect of antifibrotic therapy on survival in patients with idiopathic pulmonary fibrosis. Clin Ther. 2023;45(4):306-315. doi:10.1016/j.clinthera.2023.03.003
57. Zheng Q, Otahal P, Cox IA, et al. The influence of immortal time bias in observational studies examining associations of antifibrotic therapy with survival in idiopathic pulmonary fibrosis: a simulation study. Front Med (Lausanne). 2023:10:1157706. doi:10.3389/fmed.2023.1157706
58. Dempsey TM, Thao V, Helfinstine DA Jr, et al. Real-world cohort evaluation of the impact of the antifibrotics in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2023;62(4):2301299. doi:10.1183/13993003.01299-2023
59. Karimi-Shah BA, Chowdhury BA. Forced vital capacity in idiopathic pulmonary fibrosis—FDA review of pirfenidone and nintedanib. N Engl J Med. 2015;372(13):1189-1191. doi:10.1056/NEJMp1500526
60. Paterniti MO, Bi Y, Rekic_ D, et al. Acute exacerbation and decline in forced vital capacity are associated with increased mortality in idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2017;14(9):1395-1402. doi:10.1513/AnnalsATS.201606-458OC
61. Maher TM, Stowasser S, Voss F, et al. Decline in forced vital capacity as a surrogate for mortality in patients with pulmonary fibrosis. Respirology. 2023;28(12):1147-1153. doi:10.1111/resp.14579
62. Leard LE, Holm AM, Valapour M, et al. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379. doi:10.1016/j.healun.2021.07.005
63. Organ Procurement & Transplantation Network (OPTN) database. US Department of Health & Human Services. View data reports. Accessed October 25, 2023. https://optn.transplant.hrsa.gov/data/view-data-reports/
64. King CS, White E, Aryal S, et al. Factors associated with listing for lung transplantation in IPF patients: an analysis of the Pulmonary Fibrosis Foundation Registry. Heliyon. 2023;9(8):e18618. doi:10.1016/j.heliyon.2023.e18618
65. Goobie GC, Ryerson CJ, Johannson KA, et al. Neighborhood-level disadvantage impacts on patients with fibrotic interstitial lung disease. Am J Respir Crit Care Med. 2022;205(4):459-467. doi:10.1164/rccm.202109-2065OC
66. Swaminathan A, Hellkamp AS, Neely ML, et al. Disparities in lung transplant among patients with idiopathic pulmonary fibrosis: an analysis of the IPF-PRO Registry. Ann Am Thorac Soc. 2022;19(6):981-990. doi:10.1513/AnnalsATS.202105-589OC
67. Organ Procurement & Transplantation Network (OPTN) Policy Notice. US Department of Health & Human Services. Establish continuous distribution of lungs. March 9, 2023. Accessed December 4, 2023. https://optn.transplant.hrsa.gov/media/b13dlep2/policy-notice_lung_continuous-distribution.pdf
68. Mackintosh JA, Munsif M, Ranzenbacher L, et al. Risk of anastomotic dehiscence in patients with pulmonary fibrosis transplanted while receiving anti-fibrotics: experience of the Australian Lung Transplant Collaborative. J Heart Lung Transplant. 2019;38(5):553-559. doi:10.1016/j.healun.2019.02.005
69. Combs MP, Fitzgerald LJ, Wakeam E, et al. Pretransplant antifibrotic therapy is associated with resolution of primary graft dysfunction. Ann Am Thorac Soc. 2022;19(2):335-338. doi:10.1513/AnnalsATS.202106-736RL
70. Mora Cuesta VM, Iturbe Fernández D, Aguado Ibáñez S, et al. Antifibrotics and lung transplantation: a Spanish multicentre case-controlled study. Respirology. 2022;27(12):1054-1063. doi:10.1111/resp.14352
71. Astor TL, Goldberg HJ, Snyder LD, et al. Anti-fibrotic therapy and lung transplant outcomes in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2023;17:17534666231165912. doi:10.1177/17534666231165912

