
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Pathogenesis and Presentation of ALS: Examining Reasons for Delayed Diagnosis and Identifying Opportunities for Improvement
To claim CE credit for this activity, please visit
ABSTRACT
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig disease, is a progressive, always-fatal neuromuscular disease characterized by motor neuron degeneration in the brain and spinal cord. As upper and lower motor neurons fail, inability to transmit messages to the muscles causes muscle stiffness, atrophy, and wasting. The incidence of this incurable disease is increasing in the United States, and its prognosis is grim. On average, patients survive about 3 to 5 years from symptom onset. Until recently, few risk factors were known, but some are newly emerging. About 10% of cases are related to genetic variants. Patients who develop ALS often experience diagnostic delays (10-16 months on average), and its heterogeneity contributes to that delay. Diagnosis is based primarily on clinical signs and symptoms and exclusion of other causes of motor neuron dysfunction. Reliable, accessible biomarkers are needed to aid early ALS diagnosis, differentiate from ALS-mimicking diseases, predict survival, and monitor disease progression and treatment response. Misdiagnosing ALS can have devastating consequences, including unnecessary emotional burden, delayed and/or inappropriate treatment, and undue financial burden. The grim prognosis and sure progression to death creates considerable burden and reduces quality of life for patients and caregivers.
Am J Manag Care. 2023;29(suppl 7):S104-S111. https://doi.org/10.37765/ajmc.2023.89390
Introduction
Neurons are fundamental components of the brain and nervous system, and their proper function is critical to transmission of messages and stimuli throughout the body. They are the most diverse cells in the body with hundreds of different types possessing specific message-carrying abilities.1 When neurodegenerative diseases alter this process, the effects are debilitating.
One neurodegenerative disease is amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig disease. Amyotrophic comes from the Greek language where “a” means “no,” “myo” means “muscle,” and “trophic” means “nourishment,” meaning the muscles lack nourishment until they atrophy, or waste away.2 The disease occurs in the lateral area of the spinal cord where nerve cells that signal and control muscles are found, and neuronal degeneration causes sclerosis (ie, hardening or scarring). ALS is progressive, fatal, and incurable.
Who Gets ALS?
Every 90 minutes, someone is diagnosed with ALS and someone dies from it.3 An analysis of the National ALS Registry reported an incidence of 1.5 to 1.7 cases per 100,000 between 2014 and 2016 in the United States.4 ALS cases are on the rise; studies show that prevalence rates increased from 3.7 per 100,000 in 2002 to 5.5 per 100,000 in 2017.5,6
Experts have not identified a definite cause of ALS, but genetic, environmental, and epigenetic factors may contribute. About 10% of ALS cases are familial and the remaining are considered sporadic, meaning patients’ genetic predisposition to ALS interacts with environmental influences to cause the disease.3 About 40% to 55% of patients with familial ALS and 10% of patients with sporadic ALS have a known ALS genetic variant.7 Currently, more than 50 potentially causative or disease-modifying genes have been identified since the first gene (SOD1) was discovered in 1993. C9orf72, FUS, SOD1, and TARDBP variants are the most common genetic abnormalities implicated in ALS.7
ALS disproportionately affects White persons, men, and individuals aged 60 to 79 years.4 Family history of ALS is also another nonmodifiable risk factor for the disease. Systematic reviews and observational studies have found some modifiable risk factors including exposure to toxic metals such as lead and mercury, pesticides, and solvents; electric shock; head trauma; occupation (ie, heavy manual labor); and smoking.8 Lead exposure has the most evidence as a risk factor. A study found that hobbies and occupations involving lead (eg, casting lead bullets, making stained glass with lead joints, casting or using lead fishing sinkers) were most strongly linked with ALS.8 Holding a job in mechanics, painting, or construction was also associated with increased ALS risk. Military veterans are more likely to be diagnosed with ALS than others.3
Pathophysiology
Experts consider oxidative stress, reactive oxygen species (ROS), and glutamate excitotoxicity to be the main contributing factors to ALS progression.9 Unrestricted glutamate secretion at synaptic junctions overstimulates motor neurons receiving signals. Abnormally high calcium levels accumulate, leading to peroxidation of membrane lipids, RNA and DNA damage, mitochondrial disruption, and eventual cell death. Mitochondrial damage produces ROS as a byproduct, which reacts with lipids, proteins, and DNA.9
In healthy individuals, signals from upper motor neurons (UMN) located in the motor cortex of the brain transmit to lower motor neurons (LMN) in the spinal cord and brain stem and then to muscles in the body (see Figure 1).10 UMN dysfunction—when LMN are unable to receive messages from UMN—in patients with ALS causes muscle stiffness (spasticity) and overactive reflexes, making voluntary movements slow and difficult. When muscles are unable to receive messages—otherwise known as LMN dysfunction—they begin to weaken and shrink (atrophy and wasting) and sometimes spontaneously twitch (fasciculations).10


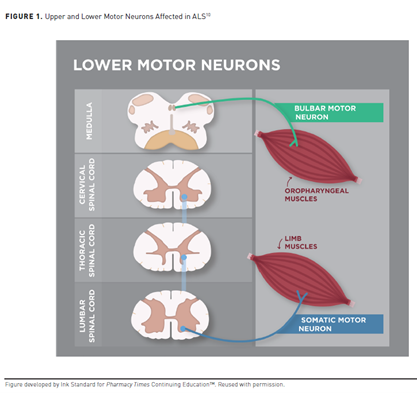
ALS pathogenesis exploits the relationship between UMN and LMN.11 Simultaneous involvement of UMN and LMN distinguishes typical ALS from atypical ALS phenotypes and other motor neuron diseases.10,12 Few ALS-mimicking conditions involve UMN and LMN clinical features, including cervical spondylotic myelopathy, spinal cysts, and spinal dural fistulas.12 Three major hypotheses discuss ALS onset11:
- Dying forward hypothesis: ALS is primarily a UMN disorder, which causes anterograde degeneration through glutamate excitotoxicity.
- Dying back hypothesis: ALS begins within the muscles or at the neuromuscular junction and noxious factors transport back from the periphery to the axon cell body to exert harmful effects.
- Independent degeneration hypothesis: UMN and LMN degeneration occurs independently.
Clinically, typical ALS spreads in a contiguous, temporal fashion.13 Disease involvement does not jump from one area to other remote areas; rather, it spreads to abutting neurons then propagates from region to region over time (see Figure 213). This also translates to symptom progression. Motor dysfunction at a focal point where symptoms initiate may progressively worsen in the same region over time, then spread outward from one side of the body to the other or from one region to the next.13
Wherever the disease originates, maximal UMN and LMN degeneration occurs.13 With contiguous spread, degeneration occurs independently at UMN and LMN levels. For example, if symptom onset begins in the right arm, LMN involvement spreads to the left arm (consistent with spinal cord anatomy), while UMN involvement spreads to the right leg (consistent with cerebral cortex anatomy).13 As the disease progresses, neuron degeneration and motor deficits become increasingly complex and ALS phenotypes become harder to differentiate.
Prognosis and Disease Burden
ALS carries a significant burden and poor prognosis.13 The average survival time is 3 years, but disease progression varies and some patients live with the disease much longer.14 About 10% of people with ALS survive 10 years, and 5% live 20 years or longer.14 Stephen Hawking, the famous physicist, cosmologist, and author, lived for more than 50 years after his ALS diagnosis. Factors that negatively affect prognosis include neurofilament light chain levels (a marker of neurodegeneration), frontotemporal dementia, changes in ALS Functional Rating Scale-Revised (ALSFRS-R), respiratory subtype, executive dysfunction, and age at onset.15
Factors that positively affect prognosis include pure-LMN or pure-UMN subtypes (vs “typical” ALS), delay in diagnosis/disease duration, and baseline ALSFRS-R score.15 Diagnostic delay and disease duration are linked because diagnostic delay reflects progression rate, meaning a longer disease course tends to have a slower progression rate. A meta-analysis found that people who had been diagnosed with ALS more than 12 months prior had a 61% lower mortality risk.15 Patients with less functional impairment at baseline also had a lower risk of death.
Presentation and Diagnosis
The initial onset of focal weakness in ALS is subtle, but it spreads relentlessly to affect most muscles.16 Disease progression is not always a straight line; some patients have periods of little-to-no loss of function lasting weeks to months.14 The disease’s heterogeneity complicates and often delays diagnosis, with devastating burden and quality of life (QOL) impact.
Clinical Presentation
Rapid motor neuron deterioration in ALS results in variable degrees of weakness, spasticity, and muscle atrophy.17 This affects key functions including limb use and ambulation, speech, and breathing. Most patients with ALS also experience dysphagia (swallowing difficulty) at some point during their disease course, and about half have some degree of frontotemporal cognitive dysfunction. Some patients also develop dementia or pseudobulbar affect (excessive emotional lability causing uncontrolled crying or laughing).17
ALS is a heterogenous journey with many different starting presentations and progression patterns but, ultimately, all patients with the disease arrive at the same destination. Most patients with ALS have “typical” presentation starting in the limbs, and about 20% and 5% experience bulbar and respiratory onset, respectively.18 While typical ALS involves both UMN and LMN, onset often predominates in one or the other. Disease onset in LMN includes spontaneous fasciculations and progressive atrophy as synapses between muscles fail. When initial deterioration occurs in UMN, patients experience brisk reflexes and slow limb coordination with muscle spasticity and stiffness.16
Ultimately, all patients will experience UMN and LMN deterioration that affects nearly all muscles.16 Fatal respiratory paralysis occurs when the disease reaches the diaphragm, usually about 3 to 5 years after disease onset.16
Atypical Subtypes of ALS
Patients with signs and symptoms of motor neuron dysfunction who do not meet true diagnostic criteria for ALS may have a related form of motor neuron disease. Atypical forms of ALS include cases with better prognosis or pure UMN or LMN involvement. These clinical phenotypes can be classified based on the level and anatomical area of motor neuron involvement and pattern of onset (see Figure 3).18 These interrelated entities and clinical syndromes usually, but not always, evolve into ALS.18


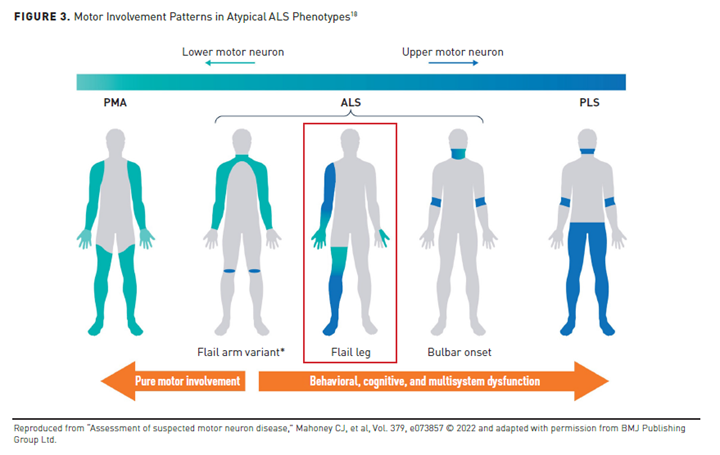
Bulbar-onset ALS is characterized by damage to the lower cranial nerves, signaled by difficulty chewing, speaking, or swallowing.16,18 Patients may also experience a brisk jaw jerk, and pseudobulbar affect is possible.Originally, clinicians considered this phenotype an entirely different disease from ALS, referring to it as “progressive bulbar palsy.”18 Symptom onset is typically gradual, but some patients become aware of problems with chewing, speaking, or swallowing quite abruptly.18 Patients with bulbar-onset ALS decline fastest among all phenotypes, survive less than 2 years from diagnosis, and have the poorest QOL.19 Poor prognosis in this subset of patients is linked to their propensity for aspiration, nutritional problems, and early respiratory involvement. Patients with bulbar-onset ALS who experience loss of ambulation have an especially poor prognosis; the mean life expectancy for these patients is just 3 months.18
Respiratory onset is most common in older and male patients as well as those with predominant LMN involvement or weight loss. While literature is inconclusive, patients with respiratory-onset ALS can be expected to have a shorter survival than patients with typical ALS given that respiratory complications are the disease’s main cause of death.20
Progressive muscular atrophy (PMA) is a syndrome limited to LMNs.13 The onset of PMA is generally later than ALS; it predominantly affects males, and it can occur in any body region. About 30% of patients develop UMN symptoms within 18 months of disease onset, and cognitive involvement is common.
Conversely, primary lateral sclerosis (PLS) syndrome is isolated to the UMN.13 Most patients experience symptom onset in the legs that ascends symmetrically to the arms and bulbar muscles. Patients with PLS and no LMN involvement 4 years post-onset often survive for decades, but LMN involvement is associated with poorer prognosis and lower survival rates.13
Flail arm or flail leg syndromes are slow-progressing phenotypes predominantly involving LMN.15 Patients with flail arm syndrome have LMN involvement restricted to the upper limbs for at least 12 months, rendering them nonfunctional, but mild UMN signs of hyperreflexia and some hypertonia are possible in the legs. Nearly all patients develop more widespread disease in less than 2 years, and the mean survival for patients with this phenotype is 4 years.15 In flail leg syndrome, LMN involvement is restricted to the lower limbs for at least 12 months and often occurs asymmetrically. After a mean of 16 months, UMN involvement emerges and the upper limbs and bulbar region are affected.15
Diagnosis
ALS diagnosis is based primarily on clinical signs and symptoms and exclusion of other causes for LMN and UMN dysfunction.17,21 Clinicians most commonly use the El Escorial criteria to diagnose ALS based on 3 factors17:
- Evidence of LMN degeneration, either on exam or through electrophysiological or neuropathological testing.
- Evidence of UMN disease on exam.
- Presence of either of the above in more than 1 region of the body.
The disease course must also be progressive with no reasonable differential diagnosis.17 Serological testing is recommended to exclude other particular diagnoses when certain clinical scenarios or presentations are seen. Common blood tests as part of investigations could include testing blood for erythrocyte sedimentation rate, C-reactive protein, a hematological screen, liver and renal function, HIV, syphilis and hepatitis screens, tests for thyroid and parathyroid diseases, vitamin B12 and folate, serum and urine protein electrophoresis, creatine kinase, and certain autoimmune antibody tests. Specialized blood tests looking for antibodies related to types of cancer (paraneoplastic panel) as well as GM-1 antibodies are performed where there is a clinical suspicion for these diagnoses. In rare cases, genetic testing to rule out ALS-mimicking diseases, including hexosaminidase-A deficiency and forms of spinal muscular atrophy, should be performed.22 In cases where there is a family history of ALS, genetic testing will typically be performed. Clinicians do not often test cerebrospinal fluid (CSF), but they can for atypical presentations or where there may be abnormalities found on spinal imaging. In those cases, CSF is examined for cell count, cytology, total protein concentration, glucose, serology (Borrelia, viral studies), and paraneoplastic antibodies. Muscle/nerve biopsies are also uncommon.23,24
Neurophysiology tests, including electromyography (EMG) and nerve conduction studies, are required for definitive ALS diagnosis.22,25 EMG evaluates the electrical activity produced by skeletal muscle to provide evidence of subclinical LMN involvement.26 Electrodes are inserted into the muscle to produce a visual display and audible signal. Nerve conduction studies assess the function and integrity of nerves in the limbs being tested by sending small electrical impulses along them and making measurements. Clinicians will also use brain, cervical, thoracic, and/or lumbar examination using magnetic resonance imaging (MRI)/computed tomography to help diagnose ALS.22,25
Table 121 lists the criteria and scoring systems used in clinical practice to diagnose, stage, and monitor ALS and its progression. Standard diagnostic criteria for ALS are lacking, which may amplify diagnostic delays, misdiagnosis, and potentially worsen prognosis.



Examining Delayed Diagnosis
Early ALS diagnosis and referral to a multidisciplinary specialized center are vital to improve outcomes, but diagnostic delays are common. An analysis of 21 retrospective studies found that the median interval from symptom onset to diagnosis for patients with ALS was between 9.1 and 27 months.17 On average, diagnosis took 10 to 16 months. Identified reasons for this delay included17:
- Patient awareness: Initial symptoms are often subtle and slow to start. Patients typically wait 3 to 6 months to seek medical attention.
- Referrals to specialists: Patients with ALS symptoms first referred to a neurologist with experience diagnosing ALS experience a shorter delay to diagnosis, but primary care providers (PCPs) often first place referrals to inappropriate specialists (eg, orthopedic surgery, neurosurgery, otolaryngology, physiotherapy, psychiatry, rheumatology).
- Misdiagnosis: In 13% to 68.4% of cases, PCPs and specialists incorrectly diagnose patients, most frequently as cerebrovascular disease, cervical myelopathy, sequelae of degenerative disc disease, radiculopathy, neuropathy, and myasthenia gravis. These patients have more advanced disease when ultimately diagnosed with ALS, and as many as 13% of incorrectly diagnosed patients also undergo unnecessary surgery prior to diagnosis.
- Site of disease onset: Patients with bulbar onset experience a shorter time to diagnosis than those with limb onset given a shorter list of differential diagnoses for bulbar-onset disease. Limb-onset symptoms may also prompt patients to visit orthopedic providers first.
- Age of onset: Almost half of patients older than 60 years are initially misdiagnosed compared with 16% of younger people.
- Neurological comorbidities: Patients with ALS who have neurological comorbidities have a diagnostic delay of 19.7 months compared with 11.1 months in patients without them. People with dementia are especially prone to these delays, with studies showing up to a 31.5-month delay from first physician contact to formal ALS diagnosis.
Variability in the ALS disease course and lack of diagnostic biomarkers makes absolute diagnosis challenging for non-ALS specialists.21 Nearly one-third of patients diagnosed with ALS do not fulfill the El Escorial diagnostic criteria for true ALS at the time of death.12 Population studies show that up to 10% of patients referred to tertiary care centers with presumptive ALS ultimately have another condition. In patients with isolated LMN features, the diagnostic error rate is close to 20%. Experts suggest that this may also stem from the fact that most ALS-mimicking diseases are even rarer than ALS itself, which even neuromuscular subspecialists may overlook. Clinical features that suggest an ALS-mimicking disease include12:
- absence of motor symptom progression
- bowel and bladder incontinence
- dysphagia out of proportion to dysarthria
- family history of another neurodegenerative disorder (except frontotemporal dementia)
- hemitongue atrophy
- multi-organ involvement outside of the neuroaxis
- multiple neurological systems involvement (eg, optic atrophy, cerebellar degeneration, seizures)
- prominent and/or early sensory and autonomic symptoms
- symptom onset early in life
In some cases, patients with true ALS present initially with multisystem involvement that is so different from classic ALS that clinicians consider them to have an entirely different disease.18,27 For example, extrapyramidal involvement or mild parkinsonism is present in up to 15% of patients with ALS and is characterized by postural instability and backward falls. Although rare in ALS, cerebellar ataxia, or damage to the cerebellum (the part of the brain controlling muscle coordination) causes clumsy voluntary movements, which can cause difficulty with walking and balance, hand coordination, speech, swallowing, and eye movements. Sensory involvement may also occur. Subjective sensory symptoms and objective sensory signs are reported in 50% and 10% of patients with ALS, respectively, but true sensory neuropathy is rare in ALS. Likewise, autonomic nervous system involvement is rare, and bladder problems are not considered a feature of ALS. However, urinary incontinence and retention are possible adverse effects of muscle relaxants and anticholinergics used for treatment of motor problems. Also, 50% to 70% of patients with PLS experience urinary urgency. Additionally, ophthalmoplegia, or paralysis of the extraocular muscles controlling eye movements, is possible in very late stages of ALS, but it is usually only seen in patients with unusually long survival and very rarely occurs shortly after disease onset.18,27
Biomarkers for ALS are challenging to access given that CSF is the main reservoir for byproducts of neuro-axonal loss.25 Although assaying low-abundance molecules in CSF is easy, retrieval is difficult. CSF testing and MRI may be reasonable in initial disease stages for early diagnosis, ruling out other diagnoses, or to determine prognosis. However, mobility loss, reduced communication skills, and frailty in end-stage disease make invasive lumbar puncture impractical. Positioning patients with clinically advanced ALS in an MRI scanner is also unrealistic given their propensity for breathing problems and pooling of secretions.25
Lack of reliable, accessible biomarkers contributes to diagnostic delays. Blood biomarkers are easily accessible, but blood is a much more complex matrix than CSF, making it more difficult to identify low-abundance molecules.25 Research has identified neurofilament elevation in the plasma and CSF as important biomarkers of disease progression, but experts have not yet incorporated them into routine care.21,25Reliable blood biomarkers would aid in early ALS diagnosis, differentiation from ALS-mimicking diseases, survival prediction, and monitoring disease progression and treatment response.
Misdiagnosing a patient with ALS can also have devastating consequences.12 It imposes emotional burden and unnecessary hardship for patients and their families and delays treatment initiation. Inappropriate treatment also leads to reduced enrollment in clinical trials and undue financial burden of obtaining disease-modifying treatments.12 The ALS Association recommends that a person diagnosed with ALS seek a second opinion from a neurologist specializing in ALS.28
Quality of Life Considerations
Functional independence and physical integrity are integral to QOL. ALS destroys these abilities.29 Functional decline associated with ALS affects patients’ ability to care for themselves, negatively impacting their health and well-being. Decreasing mobility gradually makes carrying out activities of daily living more difficult. Effects of the disease on breathing, communication, and eating are also debilitating.29
Delayed diagnosis also impacts QOL.17 Consequences of late care for ALS include severe limitations, such as dysphonia, dysarthria, and dysphagia, which can lead to gagging and bronchoaspiration.29 Noninvasive ventilation for patients with respiratory impairment improves survival and QOL, as does symptomatic treatment as ALS symptoms become prominent and incapacitating.22 Speech difficulties commonly promote emotional decline. Importantly, providers should discuss advanced directives for palliative end-of-life care proactively, respecting patients’ social and cultural backgrounds, in case communication loss occurs.22
Patients’ gradual loss of independence has a dire and remarkable impact on families’ and caregivers’ QOL.22,29 ALS progressively increases the time devoted to caring and places additional personal and social restrictions on caregivers. It contributes to psychological and emotional problems in caregivers, including depression and anxiety.22,29 Many patients lose communication skills, leading their family and caregivers to become intellectually and emotionally isolated.22
Major disparities exist among patients with ALS. One study found that Black patients have younger age at symptom onset, lower median income, longer diagnostic delay, and lower functional and ventilatory status at first clinic visit compared with White patients.30 This suggests Black patients have barriers to tertiary care. Black patients also had longer median survival time than White patients, but researchers found that race did not predict survival time independently when controlling for other variables.30
Conclusions
A quote from the novel Tuesdays with Morrie: An Old Man, a Young Man, and Life’s Greatest Lesson, summarizes the burden of ALS: “ALS is like a lit candle: it melts your nerves and leaves your body a pile of wax. You cannot support yourself standing. You cannot sit up straight. By the end, if you are still alive, your soul, perfectly awake, is imprisoned inside a limp husk. Like something from a science fiction movie, the man frozen inside his own flesh.”31 Pharmacists should appreciate the burden that this disease inflicts on patients and caregivers and the need for accurate and timely diagnosis and treatment. Understanding the variability and differing phenotypes in ALS as well as increasing familiarity with biomarkers can support definitive diagnoses of ALS in patients, in turn, leading to downstream effects of positively impacting patient QOL and survival.
Author affiliation: Karen Lynch, MD, MRCPI, is a physician and neurologist at Neurologists of Cape Cod, Cape Cod Healthcare, in Hyannis, MA.
Funding source: This activity is supported by educational grants from Amylyx and Mitsubishi Tanabe Pharma America, Inc.
Author disclosure: Dr Lynch has the following relevant financial relationships with commercial interests to disclose: Consultant: AbbVie; Speakers Bureau: AbbVie.
Authorship information: Acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, statistical analysis, and supervision.
Address correspondence to: kmlynch@capecodhealth.org
Medical writing and editorial support provided by: Kelsey Giara, PharmD, RPh
REFERENCES
- National Institute of Neurological Disorders and Stroke (NINDS). Brain basics: the life and death of a neuron. Updated November 14, 2022. Accessed January 12, 2023. www.ninds.nih.gov/health-information/public-education/brain-basics/brain-basics-life-and-death-neuron
- ALS Association. Understanding ALS. Accessed January 12, 2023. www.als.org/understanding-
- ALS Association. Who gets ALS? Accessed January 12, 2023. www.als.org/understanding-als/who-gets-als
- Mehta P, Raymond J, Punjani R, et al. Incidence of amyotrophic lateral sclerosis in the United States, 2014-2016. Amyotroph Lateral Scler Frontotemporal Degener. 2022;23(5-6):378-382. doi:10.1080/21678421.2021.2023190
- Nelson LM, Topol B, Kaye W, et al. Estimation of the prevalence of amyotrophic lateral sclerosis in the United States using national administrative healthcare data from 2002 to 2004 and capture-recapture methodology. Neuroepidemiology. 2018;51(3-4):149-157. doi:10.1159/000488798
- Mehta P, Raymond J, Punjani R, et al. Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(1-2):108-116. doi:10.1080/21678421.2022.2059380
- Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. 2019;13:1310. doi:10.3389/fnins.2019.01310
- Andrew AS, Bradley WG, Peipert D, et al. Risk factors for amyotrophic lateral sclerosis: a regional United States case-control study. Muscle Nerve. 2021;63(1):52-59. doi:10.1002/mus.27085
- Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39(2):733-748. doi:10.1002/med.21528
- National Institute of Neurological Disorders and Stroke (NINDS). Motor neuron diseases. Updated January 23, 2023. Accessed March 16, 2023. www.ninds.nih.gov/health-information/disorders/motor-neuron-diseases
- van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S. Pathophysiology and diagnosis of ALS: insights from advances in neurophysiological techniques. Int J Mol Sci. 2019;20(11):2818. doi:10.3390/ijms20112818
- Kwan J, Vullaganti M. Amyotrophic lateral sclerosis mimics. Muscle Nerve. 2022;66(3):240-252. doi:10.1002/mus.27567
- Grad LI, Rouleau GA, Ravits J, Cashman NR. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb Perspect Med. 2017;7(8):a024117. doi:10.1101/cshperspect.a024117
- ALS Association. Stages of ALS. Accessed January 19, 2023. www.als.org/understanding-als/
- Su WM, Cheng YF, Jiang Z, et al. Predictors of survival in patients with amyotrophic lateral sclerosis: a large meta-analysis. EBioMedicine. 2021;74:103732. doi:10.1016/j.ebiom.2021.103732
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162-172. doi:10.1056/NEJMra1603471
- Richards D, Morren JA, Pioro EP. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J Neurol Sci. 2020;417:117054. doi:10.1016/j.jns.2020.117054
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661-670. doi:10.1038/nrneurol.2014.184
- Shellikeri S, Karthikeyan V, Martino R, et al. The neuropathological signature of bulbar-onset ALS: a systematic review. Neurosci Biobehav Rev. 2017;75:378-392. doi:10.1016/j.neubiorev.2017.01.045
- Pinto S, Gromicho M, Oliveira Santos MO, Swash M, De Carvalho M. Respiratory onset in amyotrophic lateral sclerosis: clinical features and spreading pattern. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(1-2):40-44. doi:10.1080/21678421.2022.2067777
- Genge A, Chio A. The future of ALS diagnosis and staging: where do we go from here? Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(3-4):165-174. doi:10.1080/21678421.2022.2150555
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360-375. doi:10.1111/j.1468-1331.2011.03501.x
- Quinn C, Elman L. Amyotrophic lateral sclerosis and other motor neuron diseases. Continuum (Minneap Minn). 2020;26(5):1323-1347. doi:10.1212/CON.0000000000000911
- Šteˇtkárˇová I, Ehler E. Diagnostics of amyotrophic lateral sclerosis: up to date. Diagnostics (Basel). 2021;11(2):231. doi:10.3390/diagnostics11020231
- Sturmey E, Malaspina A. Blood biomarkers in ALS: challenges, applications and novel frontiers. Acta Neurol Scand. 2022;146(4):375-388. doi:10.1111/ane.13698
- Turner MR; UK MND Clinical Studies Group. Diagnosing ALS: the Gold Coast criteria and the role of EMG. Pract Neurol. 2022;22(3):176-178. doi:10.1136/practneurol-2021-003256
- Khemani P. Overview of adult onset cerebellar ataxia. Practical Neurology. March/April 2013. Accessed March 16, 2023. practicalneurology.com/articles/2013-mar-apr/overview-of-adult-onset-cerebellar-ataxia
- ALS Association. ALS symptoms and diagnosis. Accessed May 22, 2023. www.als.org/understanding-als/symptoms-diagnosis
- Rosa Silva JP, Santiago Júnior JB, Dos Santos EL, de Carvalho FO, de França Costa IMP, Mendonça DMF. Quality of life and functional independence in amyotrophic lateral sclerosis: a systematic review. Neurosci Biobehav Rev. 2020;111:1-11. doi:10.1016/j.neubiorev.2019.12.032
- Brand D, Polak M, Glass JD, Fournier CN. Comparison of phenotypic characteristics and prognosis between Black and White patients in a tertiary ALS clinic. Neurology. 2021;96(6):e840-e844. doi:10.1212/WNL.0000000000011396
- Albom M. Tuesdays with Morrie: An Old Man, a Young Man, and Life’s Greatest Lesson. New York, NY: Broadway Books; 2002.

