
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Optimizing Pharmacologic Treatment for ALS to Improve Outcomes and Quality of Life
To claim CE credit for this activity, please visit
ABSTRACT
Just 3 disease-modifying treatments—edaravone, riluzole, and sodium phenylbutyrate and taurursodiol (PB/TURSO)—are currently FDA approved to slow progression of amyotrophic lateral sclerosis (ALS). A fourth therapy has been recently approved under accelerated approval and is contingent upon verification of clinical benefit in confirmatory trials(s). Therapy selection is based largely upon patient characteristics, as guidelines have not been updated since the recent approval of PB/TURSO or accelerated approval of tofersen. Managing ALS symptomatically is important to improve patients’ quality of life. Although evidence is lacking for many pharmacologic therapies, providers use symptomatic treatments to address common symptoms including anxiety, depression, emotional lability (pseudobulbar affect), fasciculations, fatigue, insomnia, muscle cramps or spasms, musculoskeletal pain due to immobility, neuropathic type pain, excessive salivation (sialorrhea), spasticity, constipation, and urinary urgency. Emerging agents offer some hope for patients with ALS. Among the drugs, biologics, and interventions under investigation for ALS are an oral tyrosine kinase inhibitor, RIPK1 inhibition, the use of mesenchymal stem cells, antisense oligonucleotides, sequential administration of all experimental treatments in a new study design, and modification of the patient’s own mesenchymal stem cells.
Am J Manag Care. 2023;29(suppl 7):S112-S119. https://doi.org/10.37765/ajmc.2023.89389
Introduction
Amyotrophic lateral sclerosis (ALS) is a debilitating, progressive motor neuron disease that carries significant burden and a grim prognosis.1 ALS pathogenesis involves dysfunction of both upper motor neurons (UMN) and lower motor neurons (LMN).2 In healthy people, motor neurons form a sort of “assembly line” in the body; UMN in the brain’s motor cortex transmit signals to LMN in the spinal cord and brain stem, which propagate to muscles throughout the body.3 UMN dysfunction (inability of LMN to receive UMN signals) causes muscle stiffness (spasticity) and overactive reflexes, leading to slow and difficult voluntary movements. LMN dysfunction (inability of muscles to receive LMN signals) causes muscle atrophy and wasting as well as fasciculations (spontaneous twitching) at times.3
Most patients present with symptoms in the limbs, with about 20% and 5% experiencing bulbar and respiratory onset, respectively.4 “Typical” ALS involves UMN and LMN, though onset often predominates in one or the other. Initial onset in UMN presents as brisk reflexes and slow limb coordination with muscle spasticity and stiffness. Disease onset in LMN causes spontaneous fasciculations and progressive atrophy as synapses between muscles fail. Ultimately, all patients will experience UMN and LMN deterioration that affects nearly all muscles. About 3 to 5 years after initial onset, the disease reaches the diaphragm causing fatal respiratory paralysis.5
Initial ALS symptoms may be subtle, but the disease spreads relentlessly.5 This progression is not always constant; some patients have weeks or months with little-to-no function loss. Patients survive about 3 years on average following symptom onset, though disease progression varies and some patients live much longer.6 About 10% of people with ALS survive 10 years, and 5% live 20 years or longer.6
Curative treatment for ALS is nonexistent, but some therapies modestly slow disease progression or delay death.7 While neuroprotective therapies have limited benefit, symptomatic treatments have a greater impact on survival and quality of life (QOL).
Important principles for managing patients with ALS include respect for patient autonomy. This may be achieved by clinicians through acknowledgment ofpatients’ cultural and psychosocial beliefs and circumstances, attention to appropriate timing for decision making and recognition that goals may change over the disease course, or addressing the full continuum of care from diagnosis through palliative care.8
Treatment Goals and How to Meet Them
The American Academy of Neurology (AAN) has identified goals and desired outcomes for patients with ALS, including9:
- coordinating care among multiple providers
- enhancing QOL
- slowing disease progression
- minimizing symptoms
- educating patients and promoting patient-centered decision making
- avoiding emergent decision making by promoting advanced planning
- reducing hospitalizations
Providers use AAN and European Federation of Neurological Sciences (EFNS) guidelines in clinical practice to manage patients with ALS.9 AAN most recently updated their 2009 practice parameters for the management of ALS in January 2020.10,11 EFNS last updated their guidelines on the management of ALS in 2012.12
All guidelines stress the importance of multidisciplinary care for patients with ALS.9,11,12 Multidisciplinary care centers play a critical role in coordinating pulmonary, nutritional, and symptomatic therapies.7 In specialized multidisciplinary clinics, patients receive comprehensive care from physicians, physical therapists, occupational therapists, speech pathologists, dietitians, social workers, respiratory therapists, and nurse case managers. Multidisciplinary care affords patients various benefits. A recent review found that patients receiving care at specialized ALS clinics experience increased use of adaptive equipment; are more likely to receive prescriptions for the use of riluzole and undergo percutaneous endoscopic gastrostomy and noninvasive ventilation; as well as benefit from improved QOL and longer survival.11 Compared with care from general neurologists, patients with ALS receiving multidisciplinary care have a better prognosis, with survival lengthened up to 7.5 months.7
Bringing all providers together helps to identify patients’ most critical needs and act upon them sooner. Although few disease-modifying medications are approved for ALS, patients use many other medications for symptomatic management. Providers may refer patients with difficulty swallowing or maintaining adequate nutrition as the disease progresses for placement of gastric feeding tubes, which leads to adjustments relating to medication administration.13
A study conducted by a multidisciplinary ALS clinic evaluated the utility of their clinical pharmacist given the lack of literature surrounding pharmacist involvement in ALS care.13 Every patient saw the clinical pharmacist at each clinic visit (about every 3 months). Researchers found that patients took an average of 7.1 (3-15) total medications, including an average of 3.6 (0-10) prescription medications and 3.5 (0-13) nonprescription medications. The pharmacist made an average of 2 interventions per patient and educated patients on an average of 2.5 topics per interaction. The most common pharmacist interventions included initiating a new medication, ordering liver function tests to monitor therapy, assessing patient adherence to prescribed therapy, and ordering medication refills to improve patient adherence.13
Slowing Disease Progression
The ambiguity of ALS pathogenesis makes slowing disease progression incredibly challenging. Research indicates that oxidative stress, reactive oxygen species (ROS), and glutamate excitotoxicity are likely the main contributing factors to ALS progression.14 Unrestricted glutamate secretion at synaptic junctions overstimulates motor neurons. Abnormally high calcium levels accumulate, leading to peroxidation of membrane lipids, RNA and DNA damage, mitochondrial disruption, and eventual cell death. Mitochondrial damage produces ROS as a byproduct, which react with lipids, proteins, and DNA.14
Clinical trials have studied many experimental drugs to slow ALS progression in animal models with promising results, though few have translated to efficacy in humans with the disease.14 Three therapies are currently approved by the U.S. Food and Drug Administration (FDA) to slow disease progression in ALS, and these are riluzole, edaravone, sodium phenylbutyrate and taurursodiol (PB/TURSO). Tofersen, a fourth therapy, was approved under an accelerated approval. The Table15-20 lists appropriate dosing and administration for these disease-modifying therapies.


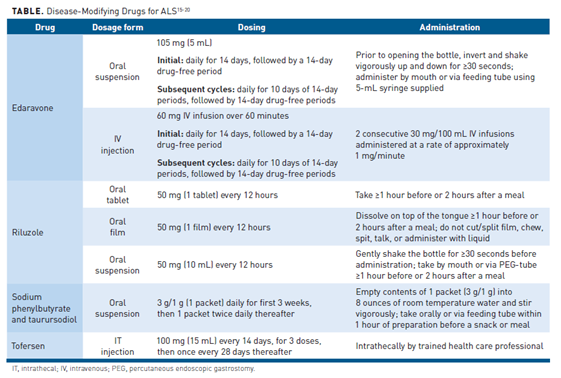
Riluzole
The mechanism of action of riluzole in ALS is poorly understood, but research suggests it inhibits glutamate reuptake in the motor neuron synapses and inactivates voltage-dependent sodium channels (ie, reduces hyperexcitability).14,17 The FDA originally approved riluzole as an oral tablet; alternative bioequivalent dosage forms include an oral suspension and a sublingual disintegrating film.21
In clinical trials, riluzole improved survival based on time to tracheostomy or death. On the functional rating scores used, the deterioration in muscle strength was slower in the riluzole group compared with placebo.22,23 Functional status was assessed with a 4-point rating that included scores for limb function, bulbar function, the results of clinical examination, and symptoms reported by the patient. A dose of 100 mg daily was associated with a 35% reduction in mortality, lengthening survival by about 3 months over a 1-year follow-up period.17,24 Recent analyses suggest that the real-world benefit of riluzole may be greater and that riluzole prolongs survival in end-stage disease (the most benefit occurs during this stage).21,24 Both AAN and EFNS guidelines state that providers should offer riluzole to all patients with ALS as early as possible after diagnosis to slow disease progression.10,12
The most frequent adverse effects (AEs) associated with riluzole are asthenia (19%), nausea (16%), decreased lung function (10%), hypertension (5%), and abdominal pain (5%).17 The drug is a cytochrome P450 (CYP) 1A2 substrate, so patients should avoid coadministration with strong or moderate CYP1A2 inhibitors (eg, ciprofloxacin, fluvoxamine, mexiletine, oral contraceptives, vemurafenib) or inducers (eg, phenytoin, rifampin, smoking).
Providers should monitor patients on riluzole for liver dysfunction (eg, elevated bilirubin), and patients with hepatic transaminase levels greater than 5 times the upper limit of normal should not take this drug.17 Some patients develop severe neutropenia within the first 2 weeks, so patients should monitor for and report signs of febrile illness. This drug also carries a warning for interstitial lung disease, including hypersensitivity pneumonitis.17
Edaravone
The mechanism of action of edaravone is also poorly understood.15 Research shows the drug acts as a ROS scavenger and inhibits peroxidation.14 The FDA first approved edaravone as an intravenous (IV) infusion in 2017 then as an oral suspension in 2022. In clinical trials, patients receiving IV edaravone had smaller declines in ALS Functional Rating Scale Revised (ALSFRS-R) scores at week 24 compared with those in the placebo group. The ALSFRS-R covers a broad range of physical functions, from speech, salivation, and swallowing to dressing, handwriting, and respiratory challenges.The scale starts at 48 points, which represents a person who is functioning normally. Individuals lose points on the scale according to loss of function based on how ALS is progressing. This scale is considered the gold standard used to measure ALS disease progression and clinical trial outcomes.25-27 Studies of edaravone oral suspension showed bloodstream drug levels comparable to the IV formulation. Additional trials are underway to determine whether higher oral edaravone doses may be beneficial.21,28
The most common AEs of edaravone are bruising (15%); gait disturbance (13%); headache (10%); dermatitis (8%); eczema (7%); and respiratory failure, respiratory disorder, or hypoxia (6%).15 In an open-label study of edaravone oral suspension, 7.6% of patients also experienced fatigue. All forms of edaravone contain sodium bisulfite, which may cause allergic reactions (including anaphylaxis and asthmatic episodes) in susceptible patients.15
Sodium Phenylbutyrate and Taurursodiol
After about 5 years without any new therapies being added to the ALS armamentarium, the FDA approved sodium phenylbutyrate and taurursodiol (PB/TURSO) in September 2022.16,21 PB/TURSO is a fixed-dose combination believed to decrease neuronal cell death by decreasing endoplasmic reticulum stress and mitochondrial dysfunction.21 Patients receiving PB/TURSO in the phase 2 CENTAUR trial (PB/TURSO, n = 89; placebo, n = 48) and its open-label extension (N = 90) had longer times to tracheostomy, permanent airway ventilation, or first hospitalization after diagnosis. There was slower disease progression as measured by ALSFRS-R in patients receiving treatment after 6 months.16 Most participants (77%) continued on a stable dose of riluzole and/or edaravone throughout the trial, and the remainder were treatment-naïve. At 35 months, the median event-free survival duration was 4.8 months longer in patients initially randomized to the treatment group compared with placebo. Median tracheostomy and permanent airway ventilation-free survival was 7.3 months longer in the PB/TURSO treatment group.29
The most common AEs of PB/TURSO are diarrhea (25%), abdominal pain (21%), nausea (18%), upper respiratory tract infection (18%), fatigue (12%), salivary hypersecretion (11%), and dizziness (10%).16 Patients with disorders impacting bile acid circulation (ie, enterohepatic circulation, pancreatic or intestinal disorders) should consult with a specialist before initiating PB/TURSO; the drug may cause or worsen diarrhea, and patients may have insufficient drug absorption. This drug’s sodium content is high (928 mg per daily maintenance dose of 2 packets); therefore sodium-sensitive patients, including those with heart failure, hypertension, or renal impairment, should be monitored appropriately.16
The choice of which therapy, combinations of therapies, or all 3 therapies and the timing of their commencement, are typically made by the treating team with the patient’s informed consent. The goal is to prolong survival and improve QOL. In some cases, clinicians may start all 3 treatments soon after each other once a diagnosis of ALS has been made; in other cases, it may be more individualized. Certain factors including how advanced the disease is, predicted life expectancy, the physical state of the patient such as ability to take medications orally, whether a G-tube is present, concomitant medical diagnoses, and respiratory involvement, may persuade a team to consider palliative approaches rather than initiate any disease-modifying treatments.
Tofersen
A new therapy with a novel mechanism of action was approved in April 2023 by the FDA through the accelerated approval process for treatment of ALS that is associated with a mutation in the superoxide dismutase 1 (SOD1) gene. Tofersen is administered intrathecally as a 100 mg/15 mL injection every 14 days for 3 doses followed by maintenance dose administered every 28 days.20 Tofersen was approved based on the results of the phase 3 VALOR clinical trial. Patients were randomized 2:1 and treated with tofersen 100 mg or placebo for 24 weeks. Concomitant riluzole and/or edaravone was permitted. The results of the study found that tofersen reduced SOD1 concentrations in cerebrospinal fluid and neurofilament light chain concentrations in plasma, though it did not improve clinical end points and was associated with AEs.30 A phase 3 open-label extension study (ATLAS) is comparing the potential effects of earlier and delayed initiation of tofersen, and results are eagerly awaited.31 Continued FDA approval is contingent upon verification of clinical benefit in confirmatory trials.
Effective Symptom Management
Although ALS is incurable, it is not untreatable. Effective symptom management is a primary goal of ALS care and key to QOL improvement.11 In addition to disease-modifying therapies, providers may prescribe medications to ameliorate various symptoms of ALS. In many cases, evidence regarding these additional therapies is limited. Unless otherwise stated, the therapies in this section of the article are used off-label.
Muscle-Related Symptoms
Muscle cramps affect more than 90% of patients with ALS.32 Although trials have assessed many agents, including mexiletine, gabapentin, vitamin E, riluzole, and quinine, for the treatment of muscle cramps, insufficient evidence exists to support or refute any pharmacologic therapy for this ALS symptom.11,32 Guidelines recommend treating cramps with physiotherapy, physical exercise, and/or hydrotherapy.9,12
Physical therapy is the mainstay of treatment for ALS-associated spasticity, but guidelines advise that providers may also try baclofen or tizanidine.9 Diazepam, baclofen, dantrolene, and tizanidine have been shown to be effective for spasticity associated with other conditions such as multiple sclerosis and cerebral palsy; however, insufficient data exist to support or refute their use in ALS.11 Research also suggests that treating spasticity may help improve gait and relieve painful spasms.
Muscle spasms can occur in any muscle in the body, including the muscles that control the opening and closing of the jaw.33 If spasms occur in these muscles, patients may have difficulty opening or closing the mouth, and a continuous or strong spasm can lead to jaw clenching. Severe jaw clenching can involve biting the sides of the tongue and the cheeks. In the event of severe, sustained, jaw-closing spasm, providers can inject botulinum toxin into the jaw-closing muscles. For cases of prolonged jaw spasticity, providers should consider baclofen, tizanidine, or benzodiazepines to relax the muscles.33
Fasciculations are common in patients with LMN dysfunction.33 They are more noticeable during periods of rest (eg, when going to bed at night). Fasciculations are not painful; many patients do not notice them until someone points them out. Generally, this symptom does not require treatment.33
Pseudobulbar Affect
About 20% to 50% of patients with ALS experience pseudobulbar affect, which is excessive laughing or crying, or involuntary emotional expression.9 These symptoms can be embarrassing and isolating, greatly diminishing QOL. Although pseudobulbar affect is not a mood disorder, providers often employ antidepressants (eg, amitriptyline, fluoxetine) to treat it.9
A combination of dextromethorphan/quinidine (DM/Q) is FDA approved to treat pseudobulbar affect.34 Patients take 1 capsule (20 mg DM/10 mg Q) orally once daily for the first 7 days, followed by 1 capsule every 12 hours thereafter. DM/Q must be used with caution as it carries many warnings and precautions. The drug increases desipramine and paroxetine exposure 8-fold and 2-fold, respectively, and may also increase digoxin substrate plasma concentration. Additionally, DM/Q is contraindicated in patients with the following conditions34:
- using quinidine, quinine, mefloquine, and other drugs that prolong QT interval, or medications metabolized by CYP2D6
- with a history of quinidine-, quinine-, or mefloquine-induced thrombocytopenia, hepatitis, or other hypersensitivity reactions
- with known hypersensitivity to dextromethorphan
- who have used a monoamine oxidase inhibitor (eg, isocarboxazid, phenelzine, selegiline, tranylcypromine) within the past 14 days
- with prolonged QT interval, congenital long QT syndrome, history suggestive of torsades de pointes, or heart failure
- with complete atrioventricular (AV) block without implanted pacemaker, or patients at high risk of complete AV block
Increased Secretions
Up to 7 of 10 patients with ALS experience sialorrhea (drooling), which is embarrassing and increases the risk of aspiration pneumonia.11 Guidelines recommend botulinum toxin B injections into the parotid and submandibular glands for patients with medically refractory sialorrhea. For patients who experience sialorrhea alternating with an uncomfortably dry mouth, atropine 0.5% or 1% eye drops administered sublingually may be a better choice given its short duration of action.12 Patients with Parkinson disease have also benefited from glycopyrrolate for sialorrhea, but there are no published trials describing its use in ALS. Transdermal scopolamine also reduces salivary flow but may cause anticholinergic AEs including confusion, urinary retention, and gastrointestinal dysmotility.12
The mucosa of the entire respiratory tract, from the nasal cavity to the lungs, contributes a constant flow of serous and mucoid fluids.12 Patients with ALS with bulbar or respiratory insufficiency are often unable to effectively clear persistent sputum. Mucus accumulation is a negative prognostic factor in patients on noninvasive ventilation, so providers should attend to this ALS symptom promptly and effectively.12 Controlled studies are lacking, but guidelines suggest that a mucolytic including N-acetylcysteine (200 mg to 400 mg 3 times daily) may be beneficial. According to the guidelines, beta-blockers (propranolol, metoprolol) and a nebulizer with saline and/or an anticholinergic bronchodilator (ipratropium, theophylline) and/or a mucolytic and/or furosemide may be used in combination; mucolytics should only be used if sufficient cough flow is present.12
Mental Health
Depression worsens QOL and shortens survival for patients with ALS.9 Prevalence varies over the disease course, but up to 44% of patients experience depression. Experts agree that depression should be treated, though there is no consensus for the best approach.11 Empirically, tricyclic antidepressants (eg, amitriptyline) and selective serotonin reuptake inhibitors (eg, escitalopram) may be effective, and mirtazapine may be better tolerated in later stages of disease.12 Anxiety is also prevalent in patients with ALS, particularly during the diagnostic and terminal phases. No systematic studies have assessed anxiolytic use in ALS, but limited evidence suggests that bupropion, oral diazepam, or sublingual lorazepam may be effective.12
Patients with ALS often experience insomnia, which may be a symptom of early respiratory weakness, pain, or underlying mental health issues (eg, anxiety, depression).9 Guidelines recommend treating insomnia with amitriptyline, mirtazapine, or appropriate hypnotics.12 However, pharmacists should note that sedative or hypnotic agents (eg, diphenhydramine, benzodiazepines, zolpidem) must be used with caution or avoided completely in patients with ALS as they could suppress respiratory drive, tidal volume, and upper airway muscle tone.9
Fatigue can be a symptom of ALS itself or an AE of medications used to slow disease progression.9 Depression, poor sleep, abnormal muscle activation, immobility, and respiratory dysfunction are all capable of causing or worsening fatigue. Guidelines do not recommend pharmacologic treatment for fatigue. However, when fatigue is a suspected AE of riluzole therapy, experts suggest that providers analyze risks of fatigue versus modest survival benefits and consider withholding the drug.11 Experts suggest that for debilitating fatigue, providers may consider modafinil.12
Additional Symptoms
Motor neuron and muscle loss are not painful processes; however, immobility due to weakness, muscle tightness, or cramping can cause pain.33 Joints that are not regularly stretched will tighten and ache, and old injuries may flare due to loss of muscle support. Patients should first try over-the-counter analgesics such as acetaminophen or nonsteroidal anti-inflammatory drugs, to reduce pain and inflammation. If these medications do not control pain sufficiently, providers may prescribe other therapies, including tramadol, opioids, or medical marijuana.33
Although ALS does not typically cause sensory nerve damage that leads to neuropathic pain (ie, tingling, burning, or electrical pain), people with ALS may have damage to these nerves inflicted by other causes.33 This type of pain can be treated with anticonvulsants (gabapentin, pregabalin), antidepressants (tricyclic antidepressants, selective norepinephrine reuptake inhibitors), and topical agents (capsaicin cream, lidocaine). Providers may also treat dyspnea and/or intractable pain as part of palliative or end-of-life care. Guidelines recommend using opioids alone or in combination with benzodiazepines if anxiety is present.12
Although literature rarely focuses on bowel and bladder symptoms of ALS, many patients experience urinary urgency and constipation. This is especially true of patients with significant UMN symptoms (ie, muscle spasticity).12 Urinary urgency causes overactive bladder wall muscles, leading to “urge incontinence.” Patients should be advised not to cut back on fluid intake to treat symptoms of incontinence as this can worsen symptoms and cause dehydration. Dehydration also causes the body to conserve water, causing constipation and making the urine more concentrated, which increases the risk of bladder lining irritation. Providers may prescribe oxybutynin (oral or transdermal) or tolterodine (oral) to suppress urinary urgency and ensure effective urinary drainage.33
Several factors may contribute to development of constipation in patients with ALS, including lack of dietary fiber, lack of mobility, increased weakness in abdominal muscles making it harder to bear down to defecate, lack of fluid intake, and AEs of medications.33 Increasing fluid intake can help if dehydration is believed to be contributing to this symptom. Several pharmacologic therapies may be used to treat constipation in patients with ALS. For example, stool softeners (docusate sodium) bring more fluid into the bowels though they may take several days to take effect. Stimulant laxatives (senna, bisacodyl, sodium picosulfate) stimulate the bowels and have been shown to take effect in about 6 to 12 hours. Glycerol suppositories act as a mild irritant to the rectum and have a quicker onset, with effect in about 15 to 60 minutes. Osmotic laxatives (magnesium salts, polyethylene glycol) increase water absorption into the stool, making it softer, bulkier, and easier to pass.33
Emerging Treatments Provide Hope
The unmet needs in ALS are immense. Fortunately, several small molecules, biologics, and interventions are in development.
Masitinib
This small molecule is an oral selective tyrosine kinase inhibitor. From studies in patients with other neurological conditions, the mechanism of action of masitinib appears to address neuroinflammation.35 Researchers propose it modulates immune mechanisms in several areas (mast cells, macrophages, and microglia); it exerts these effects in both central and peripheral nervous systems. Early placebo-controlled studies (N = 107) found that masitinib 4.5 mg/kg/day was associated with a 47% reduced risk of death over 2 years when compared with placebo.36 Functional decline also slowed.37 Outcomes were best in patients treated early (before severe functional impairment). The most frequent AEs include rash, nausea, edema, and diarrhea. Based on data indicating a dose-dependent response, a phase 3 double-blind, randomized, placebo-controlled, parallel-group study is underway to test 4.5 mg/kg/day and 6 mg/kg/day dosing.
Stem Cell Therapy
As with many diseases, the potential to use stem cells is a growing interest. Research indicates that ALS has a significant neurovascular component associated with the blood-central nervous system barrier (BCNSB), which contributes to its degenerative attributes.38 Altering the impermeability of the BCNSB impairs the neurovascular unit, so reestablishing the integrity of the BCNSB is a potential target to improve ALS prognosis. Mesenchymal stem cells (MSCs) from bone marrow or adipose tissues are being studied.
BCT-002 was a randomized, double-blind, placebo-controlled phase 3 study (N = 196) that assessed the safety and efficacy of MSC-neurotrophic factor (NTF) cells (NurOwn, autologous bone marrow-derived MSCs, induced to secrete NTFs) in participants with ALS who met revised El Escorial criteria.39 The ALSFRS-R scale was used for measurement of ALS disease progression.25,26 Researchers administered 3 intrathecal treatments of MSC-NTF or placebo. A change in disease progression of at least 1.25 ALSFRS-R points/month after treatment was considered a clinical response. Participants tolerated the treatment well, but the primary end point was not met; at 28 weeks, 33% of MSC-NTF-treated participants and 28% of placebo-treated participants met clinical response criteria (OR = 1.33; P = 0.45). Cerebrospinal biomarkers of neuroinflammation, neurodegeneration, and NTF support improved significantly in the MSC-NTF arm, while in the placebo arm these markers were unaffected. Here, too, patients with less severe disease were more likely to respond.39
The phase 3 ALSUMMIT trial (N = 115) has been designed to examine the long-term efficacy and safety of single-cycle intrathecal therapy and 3 additional booster injections of lenzumestrocel (a bone marrow stem cell product) in patients with ALS.40 In an earlier study, researchers administered 2 intrathecal treatments of autologous bone marrow-derived MSC 26 days apart. They observed therapeutic benefit lasting 6 months in patients with ALS, but it did not demonstrate long-term efficacy. Phase 3 participants will be randomized to receive 1 of 3 potential schema: a single cycle, 2 repeated injections with a 26-day interval, with placebo injections at 4, 7, and 10 months; a single cycle and 3 additional booster injections at 4, 7, and 10 months; or placebo injections at day 0, day 26, 4 months, 7 months, and 10 months (control group).
ALSUMMIT is enrolling participants with intermediate rates of ALS disease progression to increase comparison among enrollees. The primary end point will be function and survival at 6 months (study group 1 vs control) and 12 months (study group 2 vs control). These researchers are also planning a long-term follow-up observational study up to 36 months.40
Other Agents Under Development
Antisense oligonucleotides (ASOs) are another area of interest. “Fused in sarcoma” (FUS) is a predominantly nuclear DNA/RNA-binding protein that modulates RNA metabolism in cells. FUS gene variants have been linked to familial ALS (fALS-FUS).41 Although it is unclear how FUS causes toxicity, researchers are testing various hypotheses. In animal studies, young mice treated with intracerebroventricular injection of ION363, a non-allele-specific FUS ASO, had detectable levels in the central nervous system (CNS) for up to 6 months. At death, their brains and spinal cords had reduced levels of both wild-type and mutant FUS protein. These reductions are associated with delayed motor neuron degeneration. ION363 was well tolerated with no serious AEs.41 A trial of ION363 in a young woman with a FUS variant indicated no AEs and a persistence of the ASO in her CNS; however, she expired. The autopsy examination suggested some reversal in the FUS gene variant; thus, gene silencing may be a therapeutic strategy for fALS-FUS.41 A phase 3 trial is underway to determine if ION363 diffusion and action throughout the CNS with limited toxicity could improve ALS symptoms and progression; the estimated completion date is September 2025.41,42
Many serious and life-threatening diseases require multiple simultaneous approaches. The Healey ALS Platform Trial is a multicenter, multi-regimen clinical trial evaluating the safety and efficacy of multiple investigational products including zilucoplan, verdiperstat, CNM-Au8, pridopidine, and SLS-005 trehalose.43,44 It began enrollment in July 2020. Participants receive trial medications concurrently in a platform trial design following a master protocol. All investigational products are compared with placebo. At study initiation, the trial had 5 arms, but new arms will be added as new agents are identified. The researchers first randomize participants to a regimen and then randomize them to study drug or placebo in a 3:1 ratio.44 Each treatment arm is compared against a shared placebo group. The zilucoplan arm has stopped early due to futility.45
Reldesemtiv is a fast skeletal muscle troponin activator that sensitizes the sarcomere to calcium to increase muscle force.21 This oral drug is being studied in a phase 3 double-blind, randomized, placebo-controlled trial (NCT04944784; COURAGEALS). Its target enrollment is 555 participants, and its predicted closure is March 2024. Participants will be randomized (2:1) to receive 300 mg reldesemtiv twice daily or placebo. At 24 weeks, all participants will cross over to investigational reldesemtiv.21
SAR443820 is a CNS-penetrant, oral small molecule inhibitor of RIPK1, a critical signaling protein in the tumor necrosis factor receptor pathway, and a regulator of inflammation and cell death. Increased RIPK1 activity in the brain drives neuroinflammation and cell necroptosis and has been implicated in several CNS and peripheral diseases. RIPK1 inhibition has been shown to attenuate disease progression in preclinical models in ALS.46,47 A phase 2, randomized, double-blind study is assessing the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of twice daily oral SAR443820 compared with placebo and will be followed by an open-label, long-term extension study.48
Conclusions
While no cure currently exists for ALS, and disease-modifying treatments are limited, advancements in diagnosis as well as growth in the treatment armamentarium continue, and several new agents are undergoing clinical evaluation. To date, recent research and evaluation of environmental and genetic factors have linked ALS to multiple genes. Given the severity of the disease and the importance of early diagnosis and treatment to improve patient outcomes, all health care professionals, including pharmacists, must be knowledgeable and share new information and awareness with others. Increased public awareness will help improve outcomes and provide quality care to patients with ALS.
Author affiliation: Karen Lynch, MD, MRCPI, is a physician and neurologist at Neurologists of Cape Cod, Cape Cod Healthcare, in Hyannis, MA.
Funding source: This activity is supported by educational grants from Amylyx and Mitsubishi Tanabe Pharma America, Inc.
Author disclosure: Dr Lynch has the following relevant financial relationships with commercial interests to disclose: Consultant: AbbVie; Speakers Bureau: AbbVie.
Authorship information: Acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, statistical analysis, and supervision.
Address correspondence to: kmlynch@capecodhealth.org
Medical writing and editorial support provided by: Kelsey Giara, PharmD, RPh
REFERENCES
- Grad LI, Rouleau GA, Ravits J, Cashman NR. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb Perspect Med. 2017;7(8):a024117. doi:10.1101/cshperspect.a024117
- van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S. Pathophysiology and diagnosis of ALS: Insights from advances in neurophysiological techniques. Int J Mol Sci. 2019;20(11):2818. doi:10.3390/ijms20112818
- National Institute of Neurological Disorders and Stroke (NINDS). Brain basics: the life and death of a neuron. Updated November 14, 2022. Accessed January 12, 2023. www.ninds.nih.gov/health-information/public-education/brain-basics/brain-basics-life-and-death-neuron
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661-670. doi:10.1038/nrneurol.2014.184
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162-172. doi:10.1056/NEJMra1603471
- ALS Association. Stages of ALS. Accessed January 19, 2023. www.als.org/understanding-als/stages
- Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017;10:205-215. doi:10.2147/JMDH.S134992
- Andersen PM, Borasio GD, Dengler R, et al. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. an evidence-based review with good practice points. EALSC Working Group. Amyotroph Lateral Scler. 2007;8(4):195-213. doi:10.1080/17482960701262376
- American Academy of Neurology (AAN) Work Group. Amyotrophic lateral sclerosis performance measure set. Published July 31, 2012. Accessed January 20, 2023. www.aan.com/siteassets/home-page/policy-and-guidelines/quality/quality-measures/12alsmeasurementset_pg.pdf
- Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218-1226. doi:10.1212/WNL.0b013e3181bc0141
- Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227-1233. doi:10.1212/WNL.0b013e3181bc01a4
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360-375. doi:10.1111/j.1468-1331.2011.03501.x
- Jefferies KA, Bromberg MB. The role of a clinical pharmacist in a multidisciplinary amyotrophic lateral sclerosis clinic. Amyotroph Lateral Scler. 2012;13(2):233-236. doi:10.3109/17482968.2011.636449
- Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39(2):733-748. doi:10.1002/med.21528
- Radicava. Prescribing information. Mitsubishi Tanabe Pharma America, Inc.; 2022. Accessed April 30, 2023. www.radicava.com/pdfs/radicava-prescribing-information.pdf
- Relyvrio. Prescribing information. Amylyx Pharmaceuticals, Inc.; 2022. Accessed April 30, 2023. www.relyvrio.com/RELYVRIO-US-Prescribing-Information.pdf
- Rilutek. Prescribing information. Covis Pharma; 2020. Accessed April 30, 2023. www.accessdata.fda.gov/drugsatfda_docs/label/2016/020599s017lbl.pdf
- Exservan. Prescribing information. Aquestive Therapeutics; 2021. Accessed April 30, 2023. www.exservanhcp.com/assets/dist/pdfs/exservan-prescribing-information.pdf
- Tiglutik. Prescribing information. ITF Pharma, Inc.; 2020. Accessed April 30, 2023. tiglutik.com/wp-content/uploads/2020/07/TIGLUTIK_PI_with_PEG.pdf
- Qalsody. Prescribing information. Biogen, Inc; 2023. Accessed May 9, 2023. www.biogencdn.com/us/pdfs/qalsody-prescribing-information.pdf
- Johnson SA, Fang T, De Marchi F, et al. Pharmacotherapy for amyotrophic lateral sclerosis: a review of approved and upcoming agents. Drugs. 2022;82(13):1367-1388. doi:10.1007/s40265-022-01769-1
- Rozier A, Lacomblez L, Chatelier G, Bensimon G, Bouche P, Meininger V. Interrater reliability of a new rating scale for amyotrophic lateral sclerosis. J Neurol Sci. 1990;98(suppl):316-316.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585-591. doi:10.1056/NEJM199403033300901
- Fang T, Al Khleifat A, Meurgey JH, et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018;17(5):416-422. doi:10.1016/S1474-4422(18)30054-1
- Cedarbaum JM, Stambler N, Malta E, Fuller C, HiltD, Thurmond B, Nakanishi A. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. 1999;169(1-2):13-21.
- Bakker LA, Schröder CD, van Es MA, Westers P, Visser-Meily JMA, van den Berg LH. Assessment of the factorial validity and reliability of the ALSFRS-R: a revision of its measurement model. J Neurol. 2017;264(7):1413-1420. doi:10.1007/s00415-017-8538-4
- ALS trials and the ALSFRS-R. Radicava. Accessed May 1, 2023. www.radicavahcp.com/als-trials-and-alsfrs-r/
- Shimizu H, Nishimura Y, Shiide Y, et al. Evaluation of pharmacokinetics, safety, and drug-drug interactions of an oral suspension of edaravone in healthy adults. Clin Pharmacol Drug Dev. 2021;10(10):1174-1187. doi:10.1002/cpdd.925
- Paganoni S, Hendrix S, Dickson SP, et al. Effect of sodium phenylbutyrate/taurursodiol on tracheostomy/ventilation-free survival and hospitalisation in amyotrophic lateral sclerosis: long-term results from the CENTAUR trial. J Neurol Neurosurg Psychiatry. 2022;93(8):871-875. doi:10.1136/jnnp-2022-329024
- Miller TM, Cudkowicz ME, Genge A, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-1110. doi:10.1056/NEJMoa2204705
- FDA grants accelerated approval for Qalsody (tofersen) for SOD1-ALS, a major scientific advancement as the first treatment to target a genetic cause of ALS. News release. Biogen; April 25, 2023. Accessed May 1, 2023. investors.biogen.com/news-releases/news-release-details/fda-grants-accelerated-approval-qalsodytm-tofersen-sod1-als
- Oskarsson B, Moore D, Mozaffar T, et al. Mexiletine for muscle cramps in amyotrophic lateral sclerosis: a randomized, double-blind crossover trial. Muscle Nerve. 2018;10.1002/mus.26117. doi:10.1002/mus.26117
- ALS Association. Living with ALS resource guide: managing symptoms of ALS. Published 2017. Accessed February 22, 2023. www.als.org/sites/default/files/2020-04/lwals_06_2017.pdf
- Neudexta. Prescribing information. Otsuka America Pharmaceutical, Inc; 2022. Accessed May 1, 2023. www.otsuka-us.com/sites/g/files/qhldwo7066/files/media/static/NUEDEXTA-PI.pdf
- Trias E, Ibarburu S, Barreto-Núñez R, et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13(1):177. doi:10.1186/s12974-016-0620-9
- Mora JS, Bradley WG, Chaverri D, et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Ther Adv Neurol Disord. 2021;14:17562864211030365. doi:10.1177/17562864211030365
- Mora JS, Genge A, Chio A, et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1-2):5-14. doi:10.1080/21678421.2019.1632346
- Monsour M, Garbuzova-Davis S, Borlongan CV. Patching up the permeability: the role of stem cells in lessening neurovascular damage in amyotrophic lateral sclerosis. Stem Cells Transl Med. 2022;11(12):1196-1209. doi:10.1093/stcltm/szac072
- Cudkowicz ME, Lindborg SR, Goyal NA, et al. A randomized placebo-controlled phase 3 study of mesenchymal stem cells induced to secrete high levels of neurotrophic factors in amyotrophic lateral sclerosis. Muscle Nerve. 2022;65(3):291-302. doi:10.1002/mus.27472
- Nam JY, Lee TY, Kim K, et al. Efficacy and safety of lenzumestrocel (Neuronata-R inj.) in patients with amyotrophic lateral sclerosis (ALSUMMIT study): study protocol for a multicentre, randomized, double-blind, parallel-group, sham procedure-controlled, phase III trial. Trials. 2022;23(1):415. doi:10.1186/s13063-022-06327-4
- Codron P, Cassereau J, Vourc’h P. InFUSing antisense oligonucleotides for treating ALS. Trends Mol Med. 2022;28(4):253-254. doi:10.1016/j.molmed.2022.02.006
- A Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of ION363 in Amyotrophic Lateral Sclerosis Participants With Fused in Sarcoma Mutations (FUS-ALS). ClinicalTrials.gov identifier: NCT04768972. Updated February 16, 2023. Accessed March 18, 2023. clinicaltrials.gov/ct2/show/NCT04768972
- Paganoni S, Berry JD, Quintana M, et al. Adaptive platform trials to transform amyotrophic lateral sclerosis therapy development. Ann Neurol. 2022;91(2):165-175. doi:10.1002/ana.26285
- HEALEY ALS Platform Trial - Master Protocol. ClinicalTrials.gov identifier: NCT04297683. Updated December 8, 2022. Accessed February 9, 2023. clinicaltrials.gov/ct2/show/NCT04297683
- Quintanilla-Dieck M. HEALEY ALS Platform Trial Update: Zilucoplan arm stopped early for futility. Massachusetts General Hospital. March 1, 2022. Accessed February 9, 2023. www.massgeneral.org/news/press-release/healey-als-platform-trial-update-ilucoplan-arm-stopped-early-for-futility
- Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov. 2020;19(8):553-571. doi:10.1038/s41573-020-0071-y
- Wegner KW, Saleh D, Degterev A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci. 2017;38(3):202-225. doi:10.1016/j.tips.2016.12.005
- Phase 2 Study for SAR443820 in Participants With Amyotrophic Lateral Sclerosis (ALS) (HIMALAYA). ClinicalTrials.gov identifier: NCT05237284. Updated February 3, 2023. Accessed February 23, 2023. clinicaltrials.gov/ct2/show/NCT05237284

