
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Managed Care Considerations to Improve Health Care Utilization for Patients With ALS
To claim CE credit for this activity, please visit
ABSTRACT
Amyotrophic lateral sclerosis (ALS) is a fatally progressive degenerative disease, with many patients succumbing to the condition within 3 to 5 years after diagnosis. It is a rare, orphan disease with an estimated US prevalence of 25,000 patients. Patients with ALS and their caregivers are faced with a substantial financial burden as a result of the condition, as ALS has an estimated national financial burden of $1.03 billion. A significant driver of the patient financial burden includes the continued need for caregiver support as the weakening of muscles progresses to dysphagia and dyspnea, making it difficult to complete activities of daily living as the disease progresses. Caregivers also experience financial burdens, as well as feelings of anxiety, depression, and decreased quality of life. In addition to needed caregiver support, patients with ALS and their families also incur substantial nonmedical costs including travel expenses, home modifications such as ramps, and indirect costs such as productivity loss. Due to the wide range of clinical symptoms that patients may exhibit when first presenting with ALS symptoms, diagnosis is often delayed, which negatively affects patient outcomes and impacts missed opportunity for clinical trial recruitment aimed at developing new disease-modifying therapies. Additionally, delay in diagnosis and referral to ALS treatment centers results in increased overall health care costs. Telemedicine may be a tool used to promote timely care from an ALS treatment center in addition to clinical trial participation for those patients who cannot overcome mobility barriers for care. Currently, 4 therapies are approved for the treatment of ALS. Riluzole has demonstrated modest improvement in survival. Other recently approved therapies include oral edaravone, a combination therapy of sodium phenylbutyrate and taurursodiol (PB/TURSO), and tofersen, which is administered intrathecally and approved under an accelerated approval. Long-term studies have shown PB/TURSO to have a dual benefit on survivaland function. The Institute for Clinical and Economic Review (ICER) 2022 Evidence Report for ALS does not value the high price points of edaravone and PB/TURSO as cost-effective based on the current evidence, despite valuing the need for new treatment options for this patient population.
Am J Manag Care. 2023;29(suppl 7):S120-S126. https://doi.org/10.37765/ajmc.2023.89388
Introduction and Burden of Disease
Amyotrophic lateral sclerosis (ALS) is a progressive disease, with an average mortality of 3 to 5 years from diagnosis.1 ALS is more prevalent in White individuals, men, non-Hispanics, persons older than 60 years, and those with a family history.1 In 2008, the US Congress passed the ALS Registry Act, which authorized the creation of the National ALS Registry by the Centers for Disease Control and Prevention. One study that used the ALS registry, in addition to the national administrative database and additional methodologies to evaluate missing cases, estimated the prevalence of ALS in the United States in 2017 to be between 17,800 and 31,843 cases, with the mean case count to be 24,821 ALS cases (prevalence of 7.7 per 100,000 US population).1 The onset of symptoms usually occurs between the ages of 50 and 65 years.2
ALS is categorized in 2 forms as either familial or sporadic. Sporadic is the most common form, accounting for 90% to 95% of cases and with no obvious genetically inherited component. The remaining 5% to 10% are familial ALS, due to their associated genetic dominant inheritance factor. Common symptoms in both types include muscle weakness, twitching, and cramping that can eventually lead to the weakening of muscles. Advanced illness is associated with symptom progression including dyspnea and dysphagia, in addition to slurred speech, which can make it difficult to communicate with caregivers and greatly affects quality of life (QOL).3,4 The most common cause of morbidity and mortality is chronic neuromuscular respiratory failure. Patients progressively develop diaphragm weakness, causing weakened inspiratory muscle strength and the need for ventilation, weakened expiratory muscle strength needed for airway clearance, and weakened bulbar function that protects the airway from aspiration. Secretion management is required with anticholinergics and suctioning.4 In addition, patients with ALS have been shown to have higher rates of stroke and depression compared with those without ALS. Stroke, lower body mass index, and regular physical activity are risk factors for the development of ALS, although the relevance of physical activity as a risk factor has not yet been fully elucidated.5 Other potential risk factors for ALS development include family history, occupations involving manual labor, agricultural work, military service (the Gulf War in particular), football, pesticides, smoking, heavy metals, geography, and electric shock.6
Brizzi et al in collaboration with the ALS Association and a panel of experts conducted a study to evaluate the profound effects that persons with ALS and caregivers experience.7 A survey was given to patients with ALS (n = 887) and caregivers (n = 444 caregivers of living patients with ALS; n = 193 caregivers of deceased patients with ALS) to evaluate their physical and emotional symptoms, the efficacy of treatment approaches, and goals for future treatments. The most common ALS symptoms reported by both patients and caregivers were weakness and fatigue. Caregivers of deceased patients with ALS reported higher percentages of shortness of breath (72% vs 52%; P <.001), cognitive difficulty (38% vs 19%; P <.001), depression (46% vs 29%; P <.001), and difficulty sleeping (52% vs 39%; P = .004). Caregivers of both living and deceased patients reported higher stress levels than patients with ALS (P <.001). Most people living with ALS and caregivers had fears about the future, including fears of leaving family too soon, dying of respiratory failure, and spending savings on medical care, in addition to worrying about choking during the last 2 weeks of life. Of the 383 caregivers that responded to financial survey questions, 35% reported a devastating or near-devastating financial impact of ALS. When trying to seek care and find the most up-to-date, recent treatments, barriers and challenges most reported by patients and caregivers included time spent searching for care, travel to receive care, and the cost of treatments or lack of insurance coverage.7 This survey sheds light on the substantial burden of disease and the challenges patients with ALS and their caregivers face.
Cost of Care
Neuromuscular diseases are associated with significant costs to families with one or more individuals affected through direct medical costs, nonmedical costs, and indirect productivity losses through reduced household income of both the patient and caregivers. Commissioned by the Muscular Dystrophy Association, the Lewin Group aimed to measure the economic burden of ALS and its financial impact in the United States.8 The Lewin Group used commercial and Medicaid medical and pharmacy claims data to estimate the direct medical cost. Nonmedical costs were collected from a cost-of-illness patient survey developed by the researchers to account for costs such as professional caregiving services and modifications of houses and vehicles to accommodate the affected individual. Loss of productivity was also included in indirect costs and based on the “human capital” approach. This approach accounts for loss of earnings from illness or injury forgone by morbidity or mortality, and the earnings loss of household members as efforts may be directed toward the affected person’s care and away from the labor market.8
Total economic impact was calculated by multiplication of prevalence of the disease with the per-capita annual cost. Of all the neuromuscular diseases studied, ALS was associated with the highest national economic burden, totaling $1.03 billion in 2010 US dollars.8 The breakdown included $502 million attributed to direct medical costs, $287 million attributed to nonmedical costs, and $236 million attributed to indirect costs. This was the first comprehensive cost-of-illness study conducted in the United States concerning economic impact of neuromuscular disease. This study demonstrates the significant financial burden of ALS, in addition to the support needed for individuals and families affected in terms of long-term care, disease management, employment, and occupational training to improve QOL and alleviate economic burden.8
Obermann et al aimed to report annual and disease-duration costs, provide costs related to specific care and services, present payer costs, and identify strategies and resources that can be offered to patients to assist with the financial burden of ALS.9 Costs were analyzed over a 10-year period between 2001 and 2010. Total disease duration costs were estimated at $1.43 million with 85% of the costs paid by insurance, 9% paid by families, and 6% paid by charities. In-home caregivers were the highest reported costs ($669,150) followed by ventilation ($212,430) and hospital care ($114,558).9 This study further demonstrates the significant cost burden of ALS. Insurance coverage and charities can significantly reduce the cost burden to patient and families.
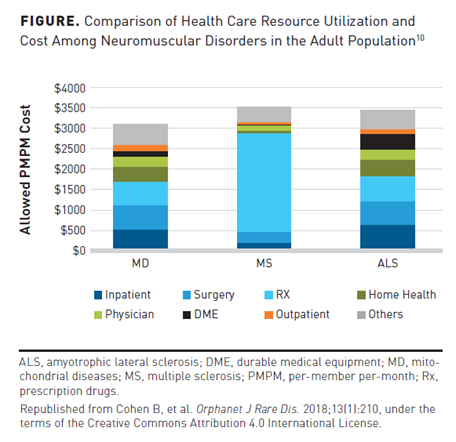
In a separate analysis evaluating the health care utilization of mitochondrial diseases, retrospective claims data were used from the Truven Health Analytics MarketScan Database and Milliman’s Consolidated Health Cost Guidelines Sources Database for the 2015 calendar year.10 Health care utilization for mitochondrial diseases was compared with that of ALS (n = 4579) and patients (n = 125,434) with multiple sclerosis. ALS was shown to have a per-member per-month (PMPM) cost burden of $3460, with most of the costs attributed to inpatient services, surgery claims, and prescription drug costs (Figure10). Inpatient visits were similar in mitochondrial diseases (27.8%) and ALS (20.9%) patient groups. Patients with multiple sclerosis were the least likely to use inpatient services (9.1%).10


A German study by Schönfelder et al aimed to estimate cost of illness depending on the clinical severity while also analyzing patients’ health-related quality of life (HRQOL).11 The study was cross-sectional and took place between July 2018 and February 2019. The revised ALS Functional Rating Scale (ALSFRS-R), a self-reported outcome scale, was used to assess disease severity. Patients were then divided into 5 groups based on King’s clinical staging system for ALS, in which stage 1 corresponded to 1 functional region involved; stage 2 to 2 regions; stage 3 to 3 regions; stage 4A to nutritional failure and stage 4B to respiratory failure; and stage 5 to death. HRQOL and health status were measured using EuroQoL (EQ-5D-5L) questionnaire; dimensions included mobility, self-care, pain/discomfort, and anxiety/depression. Patients also self-rated health on a visual analog scale (VAS; 100 = best, 0 = worst). Total cost of illness included direct medical (eg, drugs, supportive medical devices, doctors’ consultations, further therapies, and formal care) and direct nonmedical costs (eg, travel expenses, constructional alterations, legal fees, and informal care including family and nonprofessional caregivers). Indirect costs from caregivers missing work and impairment of the affected person were assessed using a human capital approach and included calculated monetary losses from reduction of work time, absent days, and early retirement. A total of 156 patients were included in the analysis, with patient age ranging from 27 years to 86 years (median age, 65 years). The mean annual cost of illness was 78,256€ per patient, with the lifetime cost per patient estimated to be 246,184€. Direct nonmedical costs accounted for the largest percentage of the cost of illness, totaling 49.1%, with informal care contributing 46.2%. Direct medical costs accounted for 35.9% of the total cost of illness, with formal care contributing 11.4% of total costs. As expected, as clinical severity increased, costs rose and QOL decreased.11 This study adds perspective to the socioeconomic burden of disease.
The ALS Association conducted the ALS Focus survey in 2020 to better understand the insurance needs and financial burdens of individuals with ALS and their families.12 The survey included 204 people living with ALS, 116 current caregivers, and 120 past caregivers (N = 440 participants). One in 4 people with ALS and caregivers said they had to borrow money or go into debt due to ALS treatment or need to provide care. One in 4 also reported they continued working beyond when they had originally planned; nearly half of those who continued working did so to maintain their health insurance coverage, either for themselves living with ALS or for the person they cared for. One in 10 reported losing their health insurance; 67% of this group reported losing their insurance because they had to stop working due to their ALS or to provide ALS care. Respondents also reported feeling significant financial stress, with a rating of 4 or 5 on a 5-point scale. The most common reasons for stress were related to medical treatments (41%) as well as receipt of medical services (39%). Financial stresses also arose from trying to understand one’s health insurance coverage (41%) and managing medical billing paperwork (38%). This focused survey shows the financial burdens and stress from insurance coverage experienced by patients with ALS and their caregivers.12 Caregiver burden was also explored by Burke et al, in which results demonstrated caregiver experiences of anxiety, depression, distress, and decreased QOL.13
Researchers aimed to better understand the determinants of QOL in ALS to address the comprehensive management of patients with ALS using the Schedule for Evaluation of Quality of Life-Direct Weighting (SEIQuL-DW) in addition to the McGill Quality of Life Questionnaire (MQoL).14 Understanding the factors related to QOL may lead to implications in the clinical setting as well as to overall patient care. Eighty patients with a mean age of 59.8 years were included with a mean duration of the disease of 2.1 years. Depression was observed in 20 patients using the Zung depression scale with scores greater than 50, 7 patients had a Beck Hopelessness Scale score over 13 demonstrating severe hopelessness, and 13 had a score between 9 and 13 demonstrating moderate hopelessness. Results demonstrated that with both QOL scales the most explicatory variable of QOL was self-perceived quality of social support. Patients did not perceive their physical status as a determinant of QOL. Furthermore, this study demonstrates the importance that physiological, supportive, and spiritual factors play when evaluating their QOL.14 Given the quick progression of disease and overall poor prognosis of ALS, maintaining QOL in patients with ALS is a complex task. To improve QOL, a focus is needed on interventions that support the family caregivers who have a heavy burden in assisting patients with ALS in their day-to-day activities.
Managed Care Considerations
Evaluating the Value of Current Treatments
There is a substantial need for new therapies for ALS. Treatment of ALS currently is aimed at supportive care that encompasses symptom management, nutritional support, and noninvasive ventilation to treat respiratory failure. Current FDA-approved therapies that modestly slow disease progression include riluzole, edaravone, PB/TURSO, and, most recently, tofersen, for patients with a mutation in the superoxide dismutase 1 (SOD1) gene. Up to the recent approval of PB/TURSO, riluzole was the only approved drug shown to improve survival (though modestly at 2-3 months) and had an A rating by the American Academy of Neurology.15 For this reason, most patients with ALS are prescribed riluzole.
Conversely, intravenous (IV) edaravone has limited use. The first treatment cycle of edaravone consists of daily infusions for 14 days, followed by a 14-day treatment-free period. Subsequent treatment cycles include daily infusions for 10 to 14 days followed by a 14-day treatment-free period. In addition to the burden of IV infusions, IV edaravone has mixed efficacy results. Edaravone may modestly slow functional loss in a subset of patients with ALS with early disease onset and slower rate of progression before randomization. For these reasons, IV edaravone use in clinical practice is limited. The FDA approved an oral suspension formulation of edaravone (Radicava ORS) based on bioequivalence with the IV formulation in May 2022. Although the dosing schedule is the same as the IV formulation, an oral formulation may overcome administration barriers, such as IV infusion risks, travel, and logistical administration concerns.16
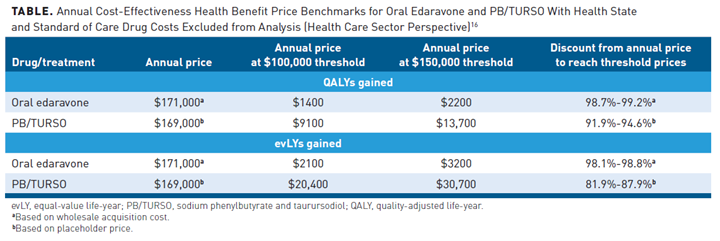
The new edaravone oral formulation has an estimated course price of $171,000 per year. Treatment with oral edaravone in addition to standard of care resulted in incremental quality-adjusted life-years (QALYs) and equal-value life-years (evLYs) of approximately 0.04 and 0.05. The incremental cost-effectiveness of oral edaravone treatment, when assuming consistent efficacy of IV therapy, far exceeds typical cost-effectiveness thresholds. The health benefit price benchmark (HBPB) for a drug is defined as the range that would achieve incremental cost-effectiveness ratios between $100,000 and $150,000 per QALY or per evLY. The ICER has rated oral edaravone a C+ for patients who meet the narrowly defined criteria (a study subgroup of early-onset ALS of the clinical trials), with an annual HBPB of $1400 to $3200.16
PB/TURSO received FDA approval in September 2022. As previously noted, up to the recent approval of PB/TURSO, only riluzole had shown a survival benefit in randomized clinical trials. However, the impact of riluzole on function in ALS is currently unclear. Long-term results from the CENTAUR trial demonstratedPB/TURSO has a dual benefit on survival and function.17,18 In addition, the findings of prolonged tracheostomy/permanent assisted ventilation-free survival and reduced hospitalization incidence supports the potential added benefits of PB/TURSO on reducing the health burden associated with ALS.19
PB/TURSO is orally administered daily for 3 weeks, then twice daily thereafter.16,20 The estimated course price of PB/TURSO is $169,000 with an annual HBPB of $9100 to $30,700. Over a lifetime, treatment of PB/TURSO in addition to standard of care showed an incremental QALY and evLYs of 0.14 and 0.31.16 The incremental cost-effectiveness of PB/TURSO would exceed typical thresholds if priced similarly to edaravone. The ICER Evidence Report for ALS rates the addition of PB/TURSO as a C++, comparable to or better than the standard of care alone. Given the need for new treatment advances, prices at the high end of typical cost-effectiveness ranges would be considered; however, these new therapies are currently above these thresholds. The ICER Report concludes that policymakers should debate short-term pricing options including a lower price close to the cost of production until further evidence is provided (Table).16


Tofersen was approved by the FDA under accelerated approval in April 2023 for the treatment of ALS in patients with a mutation in the SOD1 gene.21 The approval was based on clinical trial results showing a reduction in SOD1 concentrations in the cerebrospinal fluid and reductions in neurofilament light chain concentrations in the plasma.22 Given its recent approval within such a specific patient subgroup, how it will fit into the treatment paradigm from a financial perspective is yet to be determined.
Barriers to Care
Despite the 4 currently approved therapies for ALS, it remains a rapidly progressing, fatal disease. The median survival after symptom onset is between 3 and 5 years.2 Therefore, for early diagnosis, it is critical not only to optimize disease management and patient outcomes but also to increase opportunity for clinical trial recruitment aimed at developing new disease-modifying therapies.2 ALS can present in a wide range of clinical symptoms, and delays in diagnosis of 15 months have been reported; consequently, the mean time to arrive at a multidisciplinary ALS care clinic is 17 months.23 In a study with the purpose of characterizing diagnostic timelines and their predictors in persons with ALS, diagnosis was 51% longer in patients older than 60 years (P <.001), 46% longer in sporadic ALS compared with familial ALS (P <.043), and 45% longer in patients with limb-onset compared with bulbar-onset (P = .010) disease. Patients with symptoms such as fasciculations (46% shorter; P = .04), slurred speech (33% shorter; P = .02), and lower extremity weakness (27% shorter; P = .04) as part of the initial clinical presentation were associated with shorter ALS diagnosis.24 Patients are often referred to nonspecialist physicians and are misdiagnosed before being given their proper ALS diagnosis. The complex referral trajectory experienced by patients with ALS and their families often bypasses appropriate diagnosis and delays treatment. A fast-track system for diagnosing ALS at a specialized ALS center has been shown to reduce diagnostic delays when compared with general neurology diagnostic care pathways.25
It has been recommended to use the Gold Coast criteria in routine clinical practice when diagnosing patients.2 The Gold Coast criteria is the only diagnostic criteria that describes the minimum necessary abnormalities that would lead to a diagnosis of ALS while remaining specific and sensitive to the diagnosis. It is important to diagnose this progressive disease as early as possible given its morbidity and mortality to enable patients and families to cope with the disease and provide evidence from which to determine treatment decisions.2 In addition to better quality and timely care, earlier treatment and referral to a multidisciplinary ALS clinic has been associated with reduced cost of care by eliminating unnecessary interventions stemming from misdiagnosis.2
Insurance type may impact the coverage and access to treatment and care that is available to patients with ALS and their families. In a study aimed to describe the health care insurance and related treatment landscape among patients with ALS in the United States, data were collected from a real-world point-in-time survey of US neurologists and their consulting patients.26 Patients were grouped according to insurance coverage by Medicaid/Medicare, commercial insurance (including private or employer), and other insurance. Fifty-five neurologists included data for 372 patients with ALS. Of the patients, 55% were covered under Medicaid/Medicare, 36% under commercial health insurance, and 9% under other insurance. Patients covered under Medicaid/Medicare were prescribed fewer drugs overall. They also were less frequently prescribed disease-modifying therapies than those with commercial insurance (76% vs 83%). Symptomatic treatments including anticonvulsants and antidepressants were prescribed less frequently in the Medicare/Medicaid population than in the commercial insurance population.26
Telemedicine
Although a rare disease, the research pipeline of ALS is growing, with more than 160 companies actively involved in ALS research. There is a need to grow recruitment and trial participation as patients with ALS have limited mobility and most trials are conducted in a small number of treatment centers. Almost half of patients with ALS live more than 50 miles from a multidisciplinary ALS clinic, and one-fourth live more than 100 miles from a clinic. Many patients lack the financial means for supplies, such as vans, power chairs, and necessary personnel for safe traveling to ALS centers. Telemedicine is being used as a tool to maximize patient participation in ALS clinical trials. The COVID-19 pandemic has made the role of telemedicine more widespread for clinical care and prompted increased use in clinical trials. Telemedicine facilities’ recruitment allows information about current studies to be provided electronically and through mechanisms such as social media, search engines, and online sites and advocacy groups. Select questionnaire-based outcomes can be completed and transmitted electronically, eliminating the need for additional travel burden. Telemedicine is expected to grow to increase patient involvement, minimize patient and caregiver burden, and provide accurate and timely data for analysis in clinical trials.27
A study by Helleman et al evaluated telehealth as part of a specialized care plan for patients with ALS and the user experience of health care professionals.28 Fifty patients used ALS home-monitoring and coaching during which patients self-monitored their daily well-being, weekly weight, and monthly functional status. Telehealth services were evaluated using adoption rate (80%), dropout rate (4%), and adherence to self-monitoring; median follow-up was 11 months. Good adherence was observed in 49% of patients for well-being, 83% for body weight, and 87% for functional assessment. Most users rated telehealth as easy, helpful, not burdensome, and reported satisfaction in flexible clinic visits and continuity of care. Telemedicine via home monitoring of patients with ALS and follow-up visits with a nurse practitioner was shown to be suitable and widely accepted by patients and health care professionals in the ALS clinic setting.28 A review by Helleman et al suggested that although telehealth for ALS care is well received by patients and caregivers, barriers to adoption include reimbursement and legislation.29 Future research is needed to investigate the cost-effectiveness and appropriate reimbursement for telehealth services.
Additionally, the ICER final policy recommendations for PB/TURSO and oral edaravone for ALS publication calls for expanded access of telemedicine to deliver high-quality multidisciplinary care from specialized ALS clinics. Specialized multidisciplinary ALS clinics are the standard of care as they increase the use of evidence-based therapies, improve QOL, and may extend survival times. There are 200 ALS clinics in the United States and, of these, 73 are certified Treatment Centers of Excellence by the ALS Association. Despite the benefits of these ALS clinics, access remains a challenge. Distance and access to treatment centers may delay diagnosis, especially for minorities, low-income households, or patients living in rural areas. Telehealth is a cost-effective, feasible solution to improve access to these facilities. The COVID-19 pandemic has eased payer restrictions to telehealth services for state medical licensure, improving upon previous access issues. However, some populations may still be unable to access this service due to disparities in availability of devices, broadband connectivity, and digital health literacy. Payers and stakeholders should therefore continue to focus on improving access to telehealth and evolving accessibility to its use and growth.16
Public Health and Rare Diseases
The managed care perspective should evaluate the role of rare diseases and value-based public health. The public health approach effectively responds to sudden health crises, persistent health problems, and controlling environmental risk factors in addition to focusing on prevention and recovery. However, traditional approaches are limited in the applicability of rare disease because the diseases are extremely rare, diverse, and patients are scattered across populations. Common traits are few and far between within the category of “rare diseases.” Due to these reasons, public health approaches may not be suitable for this category because the primary measures of success are commonly preventing large numbers of cases, measuring health betterments, and avoiding premature death. However, the reasons to apply a public health approach are compelling due to the impact rare diseases have on society and the profound ability of these diseases to negatively affect the lives of patients, caregivers, and families.30
Challenges in applying value-based public health strategies include:
- Scope and capacities of registries and databases
- Lack of knowledge of most rare diseases
- Scarcity of longitudinal data collections
- Difficulty in quantifying outcomes of treatment
- Difficulty in obtaining and delayed diagnoses
- Fragmented and slow development of treatment options
- Lack of specialized and coordinated medical care
- Standards of care and rehabilitation not evidence based due to the small scale of trials and research
The standard “value-based care model” is more suited for chronic conditions such as cancer, diabetes, obesity, high blood pressure, chronic obstructive pulmonary disease, and heart disease where quantity and quality of outcomes are easier and more readily available to evaluate. Managed care organizations, stakeholders, and policy makers require that, even in the space of rare diseases, development of a new culture focuses on efficiency, efficacy, and equity at the center to radically re-examine health care. Strategies should focus on how to organize the delivery of prevention, wellness, screening, and routine health maintenance services that find ways to empower and give voices to marginalized groups such as those affected by rare diseases.30 Simply stated, value-based care moves beyond a reactive and piecemeal approach to health care to a health care strategy centered on aligning all stakeholders in the system around a common goal of doing what is right for patients. The central focus of a value-oriented health care system must be increasing value for patients, derived from measuring health outcomes against the cost of delivering the outcomes. Methods by which care pathways can be used as a tool to improve the quality of care while controlling cost are well documented in the literature. While many payers and health systems are hesitant to apply value-based arrangement to rare diseases, the reality is that rare diseases have a substantial economic impact to an already stressed health care system.
Utilization Management Considerations
The ICER final policy recommendations outline guidance for payers related to coverage and utilization management policies regarding the new treatments PB/TURSO and oral edaravone. Recommendations are summarized as follows16:
Use of prior authorizations: FDA labels should be a guide to coverage policy and should include the input of clinical experts and diverse patient representatives. Given the limited evidence and that the incremental cost-effectiveness exceeds typical cost-effective ratios for PB/TURSO and oral edaravone, prior authorizations are reasonable for payers to have on both therapies.
Minimize use of “new-to-market blocks”: Given that ALS is a rapidly progressive and fatal disease, payers should work to create formal coverage policies and evaluations before likely FDA approval dates to minimize the use of new-to-market blocks. Many payers have new-to-market blocks in place for up to 6 months after FDA approval to evaluate the evidence and for Pharmacy and Therapeutics evaluations. During this time, patients are unable to access newly approved therapies by insurance coverage. Delaying treatment by just a few months may have negative impacts on patient symptom progression, which may have been slowed with earlier treatment. Providing a path to access newly marketed therapies as soon as they are available is therefore recommended.
Coverage of ancillary home health services: The ICER recommends payers consider a benefit structure that allows patients with ALS coverage of necessary ancillary home health services such as assistive devices, home and vehicle modifications, transportation, and caregiving. Mobility issues are a significant concern for patients with ALS and their caregivers. Loss of mobility impacts performance of daily living tasks in addition to ability to attend provider and clinical trial visits. These services are frequently not covered or have inadequate coverage that results in high out-of-pocket costs and financial stress for patients and caregivers. Payers should explore benefit structures that provide support for these ancillary services.
Conclusions
ALS is a rare, progressive disease with a survival time of only 3 to 5 years after diagnosis. Given the rapid loss of muscle function often leading to ventilation and respiratory failure, timely diagnosis and treatment to slow disease progression is of paramount concern for families and patients. The cost of care is substantial, with most of the cost burden attributed to in-home caregiver support. Treatment options are limited and mainly involve supportive care, with riluzole and PB/TURSO demonstrating a benefit to survival, though only PB/TURSO demonstrates a benefit in function and reduction of hospitalizations. The clinical role of these new therapies is still to be determined, as the cost-effectiveness evaluations have shown to be lacking by the recent ICER evaluation. Therefore, managed care organizations may implement policies and procedures that support access and timely treatment for patients with ALS while balancing the incremental cost-effectiveness these new therapies provide. Managed care organizations should evaluate the access to tools such as telemedicine. Telemedicine can maximize timely care for patients by easing the care at ALS treatment centers for multidisciplinary team support and improving the ability of patients to participate in clinical trials for ALS, in addition to ancillary services that support the significant caregiver burden.
Author affiliation: Winston Wong, PharmD, is President, W-Squared Group, in Longboat Key, FL.
Funding source: This activity is supported by educational grants from Amylyx and Mitsubishi Tanabe Pharma America, Inc.
Author disclosure: Dr Wong has no relevant financial relationships with commercial interests to disclose.
Authorship information: Concept and design, critical revision of the manuscript for important intellectual content, and supervision.
Address correspondence to: wwong@wsquaredgroup.com
Medical writing and editorial support provided by: Jenna Wood, PharmD, RPh
REFERENCES
- Mehta P, Raymond J, Punjani R, et al. Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph Lateral Scler Frontotemporal Degener. 2022;24(1-2):108-116. doi:10.1080/21678421.2022.2059380
- Genge A, Chio A. The future of ALS diagnosis and staging: where do we go from here? Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(3-4):165-174. doi:10.1080/21678421.2022.2150555
- Zarei S, Carr K, Reiley L, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6:171. doi:10.4103/2152-7806.169561
- Brent JR, Franz CK, Coleman JM 3rd, Ajroud-Driss S. ALS: management problems. Neurol Clin. 2020;38(3):565-575. doi:10.1016/j.ncl.2020.03.013
- Diekmann K, Kuzma-Kozakiewicz M, Piotrkiewicz M, et al. Impact of comorbidities and co-medication on disease onset and progression in a large German ALS patient group. J Neurol. 2020;267(7):2130-2141. doi:10.1007/s00415-020-09799-z
- Hulisz D. Amyotrophic lateral sclerosis: disease state overview. Am J Manag Care. 2018;24(15 suppl):S320-S326.
- Brizzi KT, Bridges JFP, Yersak J, et al. Understanding the needs of people with ALS: a national survey of patients and caregivers. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(5-6):355-363. doi:10.1080/21678421.2020.1760889
- The Lewin Group. Cost of amyotrophic lateral sclerosis, muscular dystrophy, and spinal muscular atrophy in the United States. Muscular Dystrophy Association. Published March 1, 2012. Accessed February 23, 2023. www.mda.org/sites/default/files/Cost_Illness_Report.pdf
- Obermann M, Lyon M. Financial cost of amyotrophic lateral sclerosis: a case study. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(1-2):54-57. doi:10.3109/21678421.2014.951946
- Cohen B, Balcells C, Hotchkiss B, Aggarwal K, Karaa A. A retrospective analysis of health care utilization for patients with mitochondrial disease in the United States: 2008-2015. Orphanet J Rare Dis. 2018;13(1):210. doi:10.1186/s13023-018-0949-5
- Schönfelder E, Osmanovic A, Müschen LH, Petri S, Schreiber-Katz O. Costs of illness in amyotrophic lateral sclerosis (ALS): a cross-sectional survey in Germany. Orphanet J Rare Dis. 2020;15(1):149. doi:10.1186/s13023-020-01413-9
- ALS Association. ALS Focus results from the Understanding Insurance Needs and Financial Burdens Survey. Published 2020. Accessed February 23, 2023. www.als.org/research/als-focus/survey-results/survey-1-results
- Burke T, Galvin M, Pinto-Grau M, et al. Caregivers of patients with amyotrophic lateral sclerosis: investigating quality of life, caregiver burden, service engagement, and patient survival. J Neurol. 2017;264(5):898-904. doi:10.1007/s00415-017-8448-5
- Chiò A, Gauthier A, Montuschi A, et al. A cross sectional study on determinants of quality of life in ALS. J Neurol Neurosurg Psychiatry. 2004;75(11):1597-1601. doi:10.1136/jnnp.2003.033100
- Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218-1226. doi:10.1212/WNL.0b013e3181bc0141
- ICER publishes final evidence report and policy recommendations on treatment for amyotrophic lateral sclerosis. News release. Institute for Clinical and Economic Review; September 13, 2022. Accessed February 22, 2023. icer.org/news-insights/press-releases/icer-publishes-final-evidence-report-and-policy-recommendations-on-treatments-foramyotrophic-lateral-sclerosis/
- Paganoni S, Hendrix S, Dickson SP, et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve. 2021;63(1):31-39. doi:10.1002/mus.27091
- Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919-930. doi:10.1056/NEJMoa1916945
- Paganoni S, Hendrix S, Dickson SP, et al. Effect of sodium phenylbutyrate/taurursodiol on tracheostomy/ventilation-free survival and hospitalisation in amyotrophic lateral sclerosis: long-term results from the CENTAUR trial. J Neurol Neurosurg Psychiatry. 2022;93(8):871-875. doi:10.1136/jnnp-2022-329024
- FDA approves new treatment option for patients with ALS. News release. US FDA; September 29, 2022. Accessed February 23, 2023. www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-option-patients-als
- Qalsody. Prescribing information. Biogen, Inc; 2023. Accessed May 9, 2023. www.biogencdn.com/us/pdfs/qalsody-prescribing-information.pdf
- Miller TM, Cudkowicz ME, Genge A, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-1110. doi:10.1056/NEJMoa2204705
- Galvin M, Ryan P, Maguire S, et al. The path to specialist multidisciplinary care in amyotrophic lateral sclerosis: a population-based study of consultations, interventions and costs. PLoS One. 2017;12(6):e0179796. doi:10.1371/journal.pone.0179796
- Paganoni S, Macklin EA, Lee A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5-6):453-456. doi:10.3109/21678421.2014.903974
- Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017;10:205-215. doi:10.2147/JMDH.S134992
- Hagan M, Ciepielewska M, Mellor J, Gibson G, Earl L, Wright J. Healthcare insurance coverage and treatment patterns among patients with amyotrophic lateral sclerosis in the United States. J Manag Care Spec Pharm. Poster Abstract G9. Presented at: Academy of Managed Care Pharmacy Nexus 2021; October 2021. Accessed February 23, 2023. www.jmcp.org/doi/epdf/10.18553/jmcp.2021.27.10-b.s1
- Govindarajan R, Berry JD, Paganoni S, Pulley MT, Simmons Z. Optimizing telemedicine to facilitate amyotrophic lateral sclerosis clinical trials. Muscle Nerve. 2020;62(3):321-326. doi:10.1002/mus.26921
- Helleman J, Van Eenennaam R, Kruitwagen ET, et al. Telehealth as part of specialized ALS care: feasibility and user experiences with “ALS home-monitoring and coaching.” Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(3-4):183-192. doi:10.1080/21678421.2020.1718712
- Helleman J, Kruitwagen ET, van den Berg LH, Visser-Meily JMA, Beelen A. The current use of telehealth in ALS care and the barriers to and facilitators of implementation: a systematic review. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(3-4):167-182. doi:10.1080/21678421.2019.1706581
- Fantini B, Vaccaro CM. Value based healthcare for rare diseases: efficiency, efficacy, equity. Ann Ist Super Sanita. 2019;55(3):251-257. doi:10.4415/ANN_19_03_10

