
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Long-Chain Fatty Acid Oxidation Disorders: Managed Care and Specialty Pharmacy Implications
Abstract
Long-chain fatty acid oxidation disorders (LC-FAODs) represent a group of rare inborn errors of metabolism characterized by acute crises of energy metabolism and severe energy deficiency. Clinical manifestations include rhabdomyolysis, liver dysfunction, severe hypoglycemia, and cardiomyopathy. The symptoms associated with these genetic disorders may result in frequent hospitalizations and early death. Many patients with LC-FAODs experience major clinical events and significant morbidity and mortality that portends a substantial need for better therapeutic options for these disorders. Triheptanoin was recently approved for the treatment of LC-FAODs. As with any novel agent for rare diseases, the treatment adoption and coverage criteria for rare disease therapy such as this presents challenges for managed care decision makers. Both managed care decision makers and specialty pharmacists must ensure that healthcare professionals provide appropriate patient and caregiver education surrounding the management of LC-FAODs, including vital information on adopting emerging agents into the therapy armamentarium for these disorders.
Am J Manag Care. 2020;26:S155-S161. https://doi.org/10.37765/ajmc.2020.88479
The Burden of Long-Chain Fatty Acid Oxidation Disorders
The oxidation of fatty acids provides almost 80% of the energy required by body organs, including the heart, liver, and skeletal muscle, especially during periods of fasting when tissue glycogen stores decrease. During fasting or periods of moderate exercise or stress, the oxidation of fatty acids provides energy for cardiac and skeletal muscle while sparing glucose for use by the brain and central nervous system. Fatty acid oxidation disorders (FAODs) comprise more than 20 defects in mitochondrial β-oxidation and fatty acid transport and are inherited in an autosomal recessive pattern.1,2 Newborn testing for FAODs varies from state to state in the United States, but experience over time with this screening has demonstrated that the overall combined incidence of FAODs within the United States, Australia, and Germany encompasses a total of 5,256,999 newborns, delineated as a combined incidence of approximately 1:9300 live births.3,4
Long-chain fatty acids (LCFAs) encompass most fatty acids in both the human diet and body stores, with more than 15 enzymes involved in their oxidation processes. Genetic mutations in genes encoding these enzymes lead to long-chain fatty acid oxidation disorders (LC-FAODs), resulting in derangements in energy homeostasis. LC-FAODs are now more easily screened for after birth and many asymptomatic individuals with these disorders are identified, but many patients still develop symptoms. These symptoms appear during catabolic situations, such as fasting, leading to a heterogeneous variety of presentations, including hypoketotic hypoglycemia, liver dysfunction, rhabdomyolysis, cardiomyopathy, and even early death.5 Dietary therapy is used most frequently to manage patients with LC-FAODs, but treatment varies depending on the individual disorder and severity of the underlying enzyme deficiency. The actual effectiveness of therapy is typically reported on a case-by-case basis instead of controlled clinical trials. However, one retrospective study of 187 patients diagnosed with LC-FAODs demonstrated that overall mortality rates have not changed significantly over the past 3 decades, even with improvements in patient management. Overall, mortality remains greater than 50% in each decade, and some indications have shown higher mortality rates ranging from 60% to 95% during the review period. Newborn screening and early treatment may reduce mortality and improve outcomes but data from long-term studies are not available and patients with LC-FAODs continued to experience a major decompensation rate of about 25% despite diagnosis and standard treatment in the 2 years post birth in one study.6-8
Previous data have outlined the hospitalization burden associated with LC-FAODs. For example, data on long-term outcomes in patients with LC-FAODs demonstrated that patients averaged 1.94 hospitalizations annually for their disease, with 17.55 mean total hospital days per year. Hospitalization for rhabdomyolysis events was estimated at 1.05 events annually and hospitalization for hypoglycemia events was estimated at 0.92 events per year.6 More recent data from the triheptanoin Early Access Program (EAP) showed that among 91 patients with LC-FAODs who were critically ill before receiving triheptanoin therapy, 51% had rhabdomyolysis, 37% had cardiomyopathy, and 15% had hypoglycemia. Most patients also experienced multiple events and recurrent metabolic crises despite other treatment before administration of the protocol drug.9 Other data have linked LC-FAODs to a chronic cardiac disease phenotype and hypertrophic remodeling of the heart triggered by a mild yet chronic energy deficiency.10 These data emphasize a need for better and more individualized treatment strategies to prevent this type of decompensation that can result in hospitalization and significant morbidity and mortality.6,9
Quality of Life Impact
There is limited information regarding the impact of the symptoms associated with LC-FAODs on quality of life (QOL) in affected patients and families compared with healthy peers.11 However, various publications have revealed that patients with LC-FAODs experience substantially diminished health-related QOL (HRQOL) with negative impacts on physical, mental/emotional, and social functioning.12,13 In one study, Siddiq et al investigated the caregiver experiences of those with children with inherited metabolic diseases, its impact on child and family health, and interactions with the healthcare system.14 Investigators determined that many patients and caregivers experience increased stress and anxiety and have uncertainty and fear about the condition itself as well as long-term consequences.14 It is important to note that parents and caregivers of a chronically ill child are at greater risk of a lower HRQOL, experience more posttraumatic stress symptoms, and report higher levels of distress than parents of healthy children. Studies indicate that parental psychosocial problems affect the well-being of the child.11 In addition, although there are some limited data from early attempts at patient follow-up, more systematic monitoring of HRQOL and the psychosocial impact of LC-FAODs is needed in the future, including the functioning of patients in daily practice and comparison of their QOL to that of the general population.11
The Economic Burden of Rare Diseases and Their Management
Rare Disease Numbers and Statistics
Although the approximate incidence and prevalence of LC-FAODs has been enumerated, data surrounding the economic burden presented by these disorders are markedly lacking. An indirect comparison exploring rare disease in general can be made to assess the economic burden and costs of care associated with such disorders, including potential new therapies in development. Statistics compiled by America’s Biopharmaceutical Companies notes that more than 30 million individuals in the United States are living with some form of rare disease. Rare disease is usually defined as affecting fewer than 200,000 people in the United States and may be acute or chronic in nature. The compiled statistics outline the challenging nature of these diseases.15
Approximately 7000 rare diseases have been identified; however, just 5% of them have documented treatments available. Most of these diseases are life-threatening, emphasizing the need for new pharmacologic agents for therapy.
More than 600 orphan drugs have been approved by the FDA since the passage of the Orphan Drug Act in 1983. This is in comparison to the fact that fewer than 10 agents had been approved for rare diseases in the decade prior to this legislation.
A critical factor impacting new drug development is that 80% of rare diseases are genetic in origin. Progress in delineating these diseases at the molecular and genetic levels is crucial to drug development and advances in therapy, but it is important to note that the underlying causes of many rare diseases remain unclear.
Approximately 560 agents are currently undergoing development for the treatment of rare diseases, encompassing a variety of disorders and organ systems.
The Economic Burden of Rare Diseases: Minimal Data and Potential Frameworks for Analysis
Data surrounding the economic burden of rare diseases are lacking, which is also the case for LC-FAODs. One European analysis, the Social Economic Burden and Health-Related Quality of Life in patients with Rare Diseases in Europe study, assessed direct and indirect costs of 10 rare diseases ranging from the more common cystic fibrosis to the rarer Prader-Willi syndrome. The results showed that the cost evidence on rare diseases is very scarce. The more common rare conditions (eg, cystic fibrosis, hemophilia) were better studied, but others had very limited cost information available. Total lifetime costs were found across 4 diseases and 5 disorders. Data availability correlated with the existence of a pharmacologic therapy for the disease, and indirect costs accounted for a substantial portion of the total healthcare costs. Additionally, most of the rare diseases studied were associated with significant direct and indirect costs in terms of economic burden.16
Delineating the actual economic burden of rare disease goes beyond looking at direct and indirect costs incurred, as seen with many more common disorders. Rare diseases have fiscal impacts beyond healthcare costs. A study of the disorder hereditary transthyretin-mediated amyloidosis outlined a significant public health burden beyond the standardly measured healthcare costs. This study developed a fiscal cost model based on frameworks used to evaluate both disease burden and investments in healthcare technologies. Using a model evaluating patient income and subsequent tax payments, they assessed different scenarios based on disease onset, diagnosis, progression, and survival in terms of healthcare costs, government benefits paid to the patient, and the patient’s tax payment contributions over their lifetime. Different scenarios provided different economic results. As an example, a patient who received an early diagnosis and then exhibited median disease progression but an early death paid fewer taxes but received more in terms of pensions, disability payments, and healthcare than the general population. Such fiscal models can inform stakeholders how changes in morbidity and mortality from different patient scenarios can impact costs and expenditures, including public costs beyond the usually analyzed healthcare costs.17,18
Evaluating the Economics for Rare Disease Management: The Rise of Specialty Therapies
Over the past few years, the FDA has approved a substantial number of treatments for rare diseases. Most healthcare stakeholders want to see a continual progression of rare disease research and therapy development; however, treatment of these disorders, especially those with an underlying genetic basis, can have prohibitively high costs. These costs can make investment in research and development difficult at best, and the debate surrounding payment strategies and pricing models to address the high cost of rare disease therapy is ongoing.19
The overall global market for orphan drugs is in a period of marked acceleration. Worldwide sales of orphan drugs reached a peak of $100 billion in 2015, and that value is expected to more than double by 2022. By that point, these drugs will represent more than one-fifth of all prescription drug sales.20,21 More recent data from the annual analysis done by EvaluatePharma predicts approximately $217 billion in sales from orphan drugs in 2024.22 One key factor that drives orphan drug spending has been the higher acquisition costs associated with these agents. A recent study showed the annual costs for orphan agents to be as much as 5 times higher than for nonorphan drugs, estimated at $140,443 versus $27,756, respectively.20,21 In the United States, the use of orphan agents has been enhanced by legislative and regulatory incentives. Additionally, orphan therapies have not always been associated with barriers to insurance coverage due to the small patient population impacted and the lack of alternative therapies.20 However, while the patient populations may be small, it remains that billions of dollars are invested into performing clinical trials, regulatory approval, and marketing and commercialization of new agents for treatment of rare diseases. Payers and health systems in the United States have not actually made enough effort to understand if and how rare diseases require special considerations or how to adapt more standard methods of health technology assessment and economic evaluation to fit rare disease management. Multiple factors come into play when considering a new agent or technology, including20:
Nature of the rare disease, including impact on patients and caregivers
Impact of the new technology (eg, benefits vs risks, robustness of research evidence)
Budget impact for specialized services
Impact of the new technology beyond health benefits (eg, personal social services)
Impact of the new technology on delivery of specialized services (eg, staff training to use the new technology)
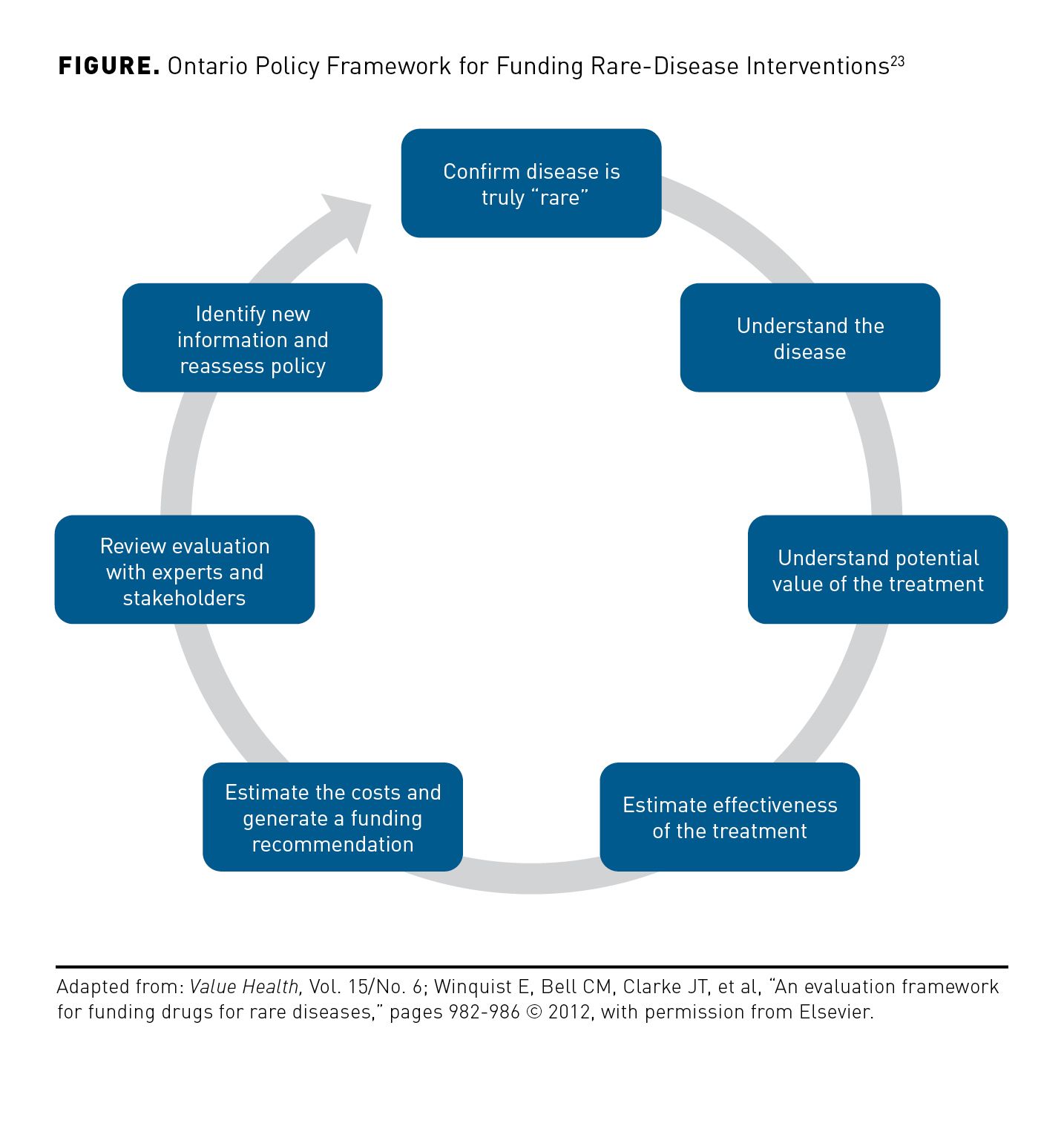
One such rare disease evaluation framework was developed in Canada for use in making decisions surrounding rare disease and associated therapy funding. The framework created by the Ontario Public Drug Program consists of 7 steps, with sustainability based on adequate evidence from clinical studies about the emerging agent, including the feasibility of clinical trials given the disease rarity and a threshold for rarity of the disorder itself (Figure23). The natural history of the disease must be reviewed along with the purported effectiveness of the new agent to fully delineate elements of the agent’s effect. Potential long-term effectiveness can then be integrated into the disease model to assess uncertainty and costs can be incorporated via a budget impact model.23-25


There is also updated information available from the Institute for Clinical and Economic Review (ICER) surrounding treatments for ultrarare diseases (defined as affecting fewer than 10,000 individuals in a population). Although they will neither change their approach to rating evidence using their own matrix nor require different standards of evidence, ICER will provide specific content surrounding potential challenges of generating evidence for treatments for these rare conditions. These may include challenges to conducting randomized clinical trials, validating surrogate outcome measures, and obtaining long-term efficacy and safety data on a particular therapy. ICER will attempt to produce a cost-effectiveness model for every new treatment that acknowledges and highlights the additional uncertainty in translating patient outcomes into the standard quality-adjusted life-year (QALY) or equal value of life gained (evLYG) measures. ICER will work to provide willingness-to-pay threshold results per QALY/evLYG over a specific range and will also investigate to find mapping studies that may allow surrogate outcomes to be translated into QOL measurement.26
Applying an Economic Model to LC-FAOD Therapy
Looking at an emerging therapy through such a lens, one can consider triheptanoin. Evaluating this new therapy through the lens of value assessment, triheptanoin is intended to provide patients with medium-length, odd-chain fatty acids that can be metabolized to increase intermediate substrates in the metabolic process. The drug can also be converted to new glucose, providing a potential additional therapeutic effect.27
Considering the early data on the effects of this agent on patient hospitalizations for LC-FAODs noted previously, triheptanoin therapy resulted in a 35% reduction in mean hospitalizations annually, decreasing from 1.94 hospitalizations per year to 1.26; P = .1126). There was also a 67% reduction in mean total hospital days annually (5.76 days vs 17.55 days before treatment; P = .0242). Hospitalization event rates for rhabdomyolysis changed from 1.05 events annually at baseline to 0.68 events per year after treatment (P = .4604) and the hypoglycemia event rate decreased by 96% (0.92 at baseline vs 0.04 following triheptanoin administration; P = .0091).6 More recent efficacy data exist from an open-label phase 2 study assessing the agent by its impact on the frequency and duration of major clinical events (eg, hospitalizations, emergency department visits, and emergency home visitations for symptoms related to LC-FAODs). Results demonstrated that triheptanoin decreased mean annualized event rates by almost half from 1.69 to 0.88 events per year (P = .021), with the mean annualized duration rate decreasing from 5.96 to 2.96 days annually (P = .028). Data such as these show the drug’s potential benefit in reducing disease burden and direct medical costs associated with symptomatic events. As a result of the efficacy and medical cost offsets, the newly approved product has an annual therapy costing approximately $138,000 per patient per year.27 It must also be noted that in April 2019, triheptanoin was granted a fast-track designation and rare pediatric disease designation by the FDA for treatment of LC-FAODs, further commenting on the potential overall value of this therapy.28 This fast-track status led to the recent FDA approval of triheptanoin in June 2020.29
Managed Care Considerations in Treatment of Rare Diseases, Including Emerging Therapies
Managed Care Providers and Patient Population Data
Health plans and payers who can provide information surrounding patient population data can also provide a more in-depth view into the management of patients with rare diseases. Rare diseases in general take longer to diagnose, incur greater rates of hospitalization, require oversight by both primary care providers and specialists, and usually require a greater volume of diagnostic tests and procedures for optimal management. In addition, rare diseases, especially those with symptoms of variable severity and appearance, may incur substantial healthcare resource utilization, complicated by uncertainty around technologies needed for diagnosis and especially for options surrounding treatment.30-32 In a managed care setting, intensive analysis of population health data surrounding a rare disease can help further educate providers to gain a better and more comprehensive understanding of symptoms, treatments, and procedures needed for optimal disease management. A clearer and more complete understanding of risk factors and treatment options for patients with rare diseases will help support health plans in optimizing the overall health of their patient population. Such an inclusive approach and data analysis could improve patient outcomes and even patient QOL while reducing the economic burden associated with rare disease.30
Managed care professionals have several considerations when determining access to orphan drugs. Necessary considerations for new agents in development and their potential future coverage and availability include33:
- Timing: when will payers be impacted?
- Clinical effectiveness: is this agent better than other available options?
- Patient experience: will this positively or negatively impact patients?
- Payer impact: will this influence prescription spend?
Addressing the High Treatment Costs for Rare Disease in the Managed Care Setting
Managed care providers and organizations continue to debate the best strategies to address the high costs of rare disease treatment to provide optimal care for patients with these conditions. One recent managed care pharmacy survey by the publication Managed Healthcare Executive asked managed care consultants, health plans, and pharmacy benefit managers to identify the best long-term approach to managing rare disease costs. Results demonstrated that 28% of participants called for a more integrated health approach by benefit managers surrounding the total cost of care across the healthcare continuum. In addition, 22% of participants called for a National Institute for Health and Care Excellence-based approach or even ICER-based limitations for some therapy access that would center on a measure of cost-benefit (eg, cost or impact on QALY). Data also showed that 19.6% of respondents suggested better assessment of treatment risk versus benefit and discontinuation when clinically appropriate. However, the National Organization for Rare Disorders (NORD) offered cautionary statements on implementing such stringent procedures. Per the vice president of Regulatory and Government Affairs at NORD, the FDA is approving a record number of orphan therapies with a corresponding increase in record prices set for these agents. Utilization management strategies are the cornerstone to controlling overall healthcare costs when multiple brand-name agents, generics, or biosimilar alternatives are available. However, many patients with rare diseases often have just 1 option for treatment. More aggressive utilization management techniques will not work in these circumstances.19 Recognizing the need for rare disease treatments, managed care organizations have begun expanding medical necessity criteria to go beyond simple physician attestation of disease alone. The shift is to require testing and rigorous documentation to support the diagnosis and confirm the medical necessity of these high-cost therapies. In some instances, organizations have moved to stringent criteria that limit therapy strictly to specific patient populations who were studied in clinical trials as opposed to standard FDA-approved indications. The goal of this shift in management approach is to better manage cost by ensuring appropriate utilization in the absence of lower cost alternative therapies.
The Role of the Specialty Pharmacist in Rare Disease Management
The Specialty Pharmacist as Part of the Interprofessional Healthcare Team
Specialty pharmacies play a critical role at the center of pharmaceutical treatments for rare diseases. Often, specialty pharmacies utilize a team of clinicians and administrative staff to manage these patients. Activities include coordination with payers through the authorization process, drug and disease education to patients, and coordination of medication delivery and administration as well as alternative coverage opportunities using patient assistance programs. This can be a period of high anxiety for the patients and those assisting in their care. Having the support from the specialty pharmacy team is critical in ensuring appropriate use of these extremely high-cost medications.34
Specialty pharmacists must work in concert with other members of the multidisciplinary and interprofessional healthcare team to help mitigate the impact of rare diseases in both patients with these disorders as well as their families and caregivers. This type of support requires precise coordination among the patient, caregivers, physicians, specialists, pharmacists, payers, and drug manufacturers. The patient-centered approach that is needed extends well beyond dispensing therapies. Specialty pharmacists must focus on all the fundamentals involved in patient management in rare diseases, especially compassionate and effective communication surrounding the administration and use of the prescribed agent, the nature of the disease itself, and any concomitant patient conditions and comorbidities. Many patients with rare diseases require multiple therapeutic agents, and the treatment regimens to manage their symptoms and prevent or limit adverse effects can be quite complex. The intensive skills of the specialty pharmacist may be needed around the clock to provide the necessary patient and caregiver education and support that enhances patient–prescriber interactions.35
Triheptanoin is supplied as an oral liquid in a 500-mL glass bottle and requires the patient or caregiver to draw appropriate doses and self-administer either orally or enterally. Specialty pharmacists can help clarify administration questions and coordinate with the healthcare team to optimize care. If a patient has difficulty tolerating a quarter of the total dose 4 times daily, the dose can be further reduced and given more frequently. If the patient experiences gastrointestinal adverse effects, the total daily dose can be reduced and more slowly increased back to target. It can be mixed with semi-solid food or liquids such as plain or artificially sweetened fat-free yogurt, fat-free milk, formula or cottage cheese, whole grain hot cereal, and fat-free low-carbohydrate pudding, smoothies, applesauce or baby food and given orally or through a feeding tube. It should be prepared and administered in compatible containers, syringes, or measuring cups made of stainless steel, glass, high-density polyethylene, polypropylene, low-density polyethylene, polyurethane, and silicone. It should not be administered using plastics made of polystyrene or polyvinyl chloride. Triheptanoin should be thoroughly mixed into the desired food or liquid and can be stored refrigerated for up to 24 hours after being mixed.29
In addition to helping patients with rare diseases with both their general and specific needs, the specialty pharmacist can increase the prescriber’s visibility into factors affecting therapy access, adherence, and outcomes by providing actionable updates to improve the overall management pathways.35 Advances in drug development in precision medicine require delicate and careful management, considering the overall costs to patients, payers, and society. In the case of rare diseases, the smaller the patient population affected, the higher likelihood of having a semi-exclusive or exclusive specialty pharmacy dispensing channel, thus allowing better data management, flexibility, and ability to identify and manage therapy results and trends in affected patients.35 As more pharmacies enter the rare disease market, they must put in place the structure and technology to assist every individual patient using these potentially complex therapies. Pharmacy programs will need to incorporate specialized teams with the ability to use program-specific technologies that will lead them through the patient management process and collect and analyze data on the use of these new agents. Overall, pharmacies must develop and implement programs that differ substantially from those for other specialty drugs to provide optimal patient management in the rare disease landscape.36 The specialty pharmacist will be at the center of managing patients and therapies that require high-touch, greater distribution control, and more real-time data. These pharmacists must help channel rare disease treatments to those who need them the most, and in their specialty setting, they are the practitioners most ideally suited to meet this need.35
Conclusions
LC-FAODs are rare diseases, and the symptoms associated with these genetic disorders may result in significant morbidity and mortality, including frequent hospitalizations and even early death. With the potential for a novel agent on the horizon to treat LC-FAODs, new challenges in managing both the clinical and economic burdens of these disorders will arise for managed care decision makers and specialty pharmacists. These healthcare professionals must thoroughly assess emerging agents for therapy in terms of efficacy, safety, and cost-effectiveness, using evolving algorithms and pathways for determining their optimal use in individual patients. They must also provide appropriate patient and caregiver education on adopting emerging agents into therapy plans in concert with the multidisciplinary and interprofessional healthcare team to optimize patient therapy and improve outcomes in patients with LC-FAODs.
Author affiliation: Alan Pannier, PharmD, MBA, is Head of Clinical Services, SmithRx, Salt Lake City, UT.
Funding source: This activity is supported by an educational grant from Ultragenyx Pharmaceutical Inc.
Author disclosure: Dr Pannier has no relevant financial relationships with commercial interests to disclose.
Authorship information: Substantial contributions to the concept and design; drafting of the manuscript; overall supervision; and critical revision of the manuscript for important intellectual content.
Address correspondence to: alan.pannier@smithrx.com
Medical writing and editorial support: Elizabeth Paczolt, MD, FACNM
REFERENCES
1. Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57(5 Pt 2):78R-86R. doi: 10.1203/01.PDR.0000159631.63843.3E
2. Bennett MJ, Rinaldo P, Strauss AW. Inborn errors of mitochondrial fatty acid oxidation. Crit Rev Clin Lab Sci. 2000;37(1):1-44. doi: 10.1080/10408360091174169
3. Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation:
experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33(5):521-526. doi: 10.1007/s10545-010-9076-8
4. Conditions screened by state. Baby’s First Test. Accessed June 16, 2020. babysfirsttest.org/newborn-screening/states
5. Knottnerus SJ, Bleeker JC, Wüst RC, et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endoc Metab Disord. 2018;19(1):93-106. doi: 10.1007/s11154-018-9448-1
6. Vockley J, Marsden D, McCracken E, et al. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment—a retrospective chart review. Mol Genet Metab. 2015;116(1-2):53-60. doi: 10.1016/j.ymgme.2015.06.006
7. Baruteau J, Sachs P, Broue P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013;36(5):795-803. doi: 10.1007/s10545-012-9542-6
8. Lindner M, Gramer G, Haege G, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases—report of 10 years from south-west Germany. Orphanet J Rare Dis. 2011;6:44. doi: 10.1186/1750-1172-6-44
9. Bedrosian SJ, Marsden D, Asha S, Kruger E. Mortality associated with long-chain fatty acid oxidation disorders: observations from an expanded access program for triheptanoin. J Manag Care Pharm. 2020;26(4-a):S30. Abstract E28.
10. Knottnerus SJ, Bleeker JC, Ferdinandusse S, et al. Subclinical effects of long-chain fatty acid β-oxidation deficiency on the adult heart: a case-control magnetic resonance study. J Inherit Metab Dis. Published online May 28, 2020. doi: 10.1002/jimd.12266
11. Knottnerus SJ, Bleeker JC, van Hasselt PM, Wijburg FA, Visser G. Health-related quality of life in patients with long-chain fatty acid oxidation disorders. Published December 26, 2018. Accessed June 16, 2020. informnetwork.org/2018/12/26/health-related-quality-of-life-in-patients-with-long-chain-fatty-acid-oxidation-disorders/
12. Vockley J, Burton B, Berry GT, et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: safety and efficacy in children and adults following 24 weeks of treatment. Mol Genet Metab. 2017;120(4):370-377. doi: 10.1016/j.ymgme.2017.02.005
13. Yamada K, Taketani T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Human Genet. 2019;46(2):73-85. doi: 10.1038/s10038-018-0527-7
14. Siddiq S, Wilson BJ, Graham ID, et al. Experiences of caregivers of children with inherited metabolic diseases: a qualitative study. Orphanet J Rare Dis. 2016;11(1):168. doi: 10.1186/s13023-016-0548-2
15. Rare diseases by the numbers. America’s Biopharmaceutical Companies. Accessed May 20, 2020. innovation.org/about-us/commitment/research-discovery/rare-disease-numbers
16. Angelis A, Tordrup D, Kanavos P. Socio-economic burden of rare diseases: a systematic review of cost of illness evidence. Health Policy. 2015;119(7):964-979. doi: 10.1016/j.healthpol.2014.12.016
17. Connolly MP, Panda S, Patris J, Hazenberg BP. Estimating the fiscal impact of rare diseases using a public economic framework: a case study applied to hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2019;14(1):220. doi: 10.1186/s13023-019-1199-x
18. Joszt L. Analysis highlights how rare diseases have broader fiscal impact than health costs. Am J Manag Care. Published October 15, 2019. Accessed May 21, 2020. ajmc.com/newsroom/analysis-highlights-how-rare-diseases-have-broader-fiscal-impact-than-health-costs
19. Sukel K. Balancing the costs of rare disease treatment. Managed Healthcare Executive. Published June 14, 2019. Accessed May 22, 2020. managedhealthcareexecutive.com/managed-care-pharmacy-survey/balancing-costs-rare-disease-treatment
20. Ollendorf DA, Chapman RH, Pearson SD. Evaluating and valuing drugs for rare conditions: no easy answers. Value Health. 2018;12(5):547-552. doi: 10.1016/j.jval.2018.01.008
21. EvaluatePharma world preview 2017, outlook to 2022. Evaluate. Published June 2017. Accessed May 23, 2020. info.evaluategroup.com/rs/607-YGS-364/images/WP17.pdf
22. EvaluatePharma Orphan Drug Report 2020. Accessed June 16, 2020. evaluate.com/orphan-drugs
23. Winquist E, Bell CM, Clarke JT, et al. An evaluation framework for funding drugs for rare diseases. Value Health. 2012;15(6):982-986. doi: 10.1016/j.jval.2012.06.009
24. Winquist E, Coyle D, Clarke JT, et al. Application of a policy framework for the public funding of drugs for rare diseases. J Gen Intern Med. 2014;29(suppl 3):S774-S779. doi: 10.1007/s11606-014-2885-y
25. Blackburn H, Labarthe D. Stories from the evolution of guidelines for causal inference in epidemiologic associations: 1953-1965. Am J Epidemiol. 2012;176(12):1071-1077. doi: 10.1093/aje/kws374
26. Modifications to the ICER value assessment framework for treatments of ultra-rare diseases. Institute for Clinical and Economic Review. Updated January 31, 2020. Accessed June 16, 2020. icer-review.org/wp-content/uploads/2020/01/ICER_URD_Framework_Adapt_013120.pdf
27. Vockley J, Burton B, Berry GT, et al. Results from a 78-week, single-arm, open-label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long-chain fatty acid oxidation disorders (LC-FAOD). J Inherit Metab Dis. 2019;42(1):169-177. doi: 10.1002/jimd.12038
28. Ultragenyx announces UX007 granted Fast Track Designation and Rare Pediatric Disease Designation by U.S. FDA for treatment of long-chain fatty acid oxidation disorders. News release. Intrado GlobeNewswire; April 16, 2019. Accessed May 24, 2020. globenewswire.com/news-release/2019/04/16/1804740/0/en/Ultragenyx-Announces-UX007-Granted-Fast-Track-Designation-and-Rare-Pediatric-Disease-Designation-by-U-S-FDA-for-Treatment-of-Long-Chain-Fatty-Acid-Oxidation-Disorders.html
29. Dojolvi. Prescribing information. Ultragenyx Pharmaceutical Inc; 2020. Accessed July 15, 2020. ultragenyx.com/medicines/dojolvi-full-prescribing-information
30. Johnson MP, Johnson JC, Engel-Nitz NM, et al. Management of a rare disease population: a model for identifying a patient population with tuberous sclerosis complex. Managed Care Magazine. Published August 28, 2017. Accessed May 24, 2020. managedcaremag.com/archives/2017/8/management-rare-disease-population-model-identifying-patient-population-tuberous
31. Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371(9629):2039-2041. doi: 10.1016/S0140-6736(08)60872-7
32. de Vrueh R, Baekelandt ER, de Haan JM. Priority Medicines for Europe and the World: “A Public Health Approach to Innovation.” Update on 2004 Background Paper. Background Paper 6.19 Rare Diseases. World Health Organization; March 12, 2013. Accessed May 24, 2020. who.int/medicines/areas/priority_medicines/BP6_19Rare.pdf
33. Orphan drug debate: understanding the increasingly popular and competitive orphan drug market while balancing this costly trend. EnvisionRx. Published July 19, 2018. Accessed May 24, 2020. visiblydifferent.envisionrx.com/blog/perspective-on-the-rx-pipeline-orphandrugs
34. Boutwell L. Ask the pharmacist: after a rare disease diagnosis, what’s next? Express Scripts. Published February 26, 2020. Accessed May 25, 2020. express-scripts.com/corporate/articles/ask-pharmacist-after-rare-disease-diagnosis-whats-next
35. Sukel K. Specialty pharmacy’s role in improving orphan drug channels. Managed Healthcare Executive. Published May 3, 2019. Accessed July 21, 2020. managedhealthcareexecutive.com/view/specialty-pharmacys-role-improving-orphan-drug-channels
36. Rare pharmacy emerges from specialty (pharmacy). PANTHERx Rare Pharmacy. Published May 4, 2020. Accessed June 17, 2020. pantherxrare.com/blog/white-paper-rare-pharmacy-emerges-from-specialty-pharmacy-published
Click here to download PDF version
