
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Long-Chain Fatty Acid Oxidation Disorders and Current Management Strategies
Abstract
Long-chain fatty acid oxidation disorders (LC-FAODs) are rare, life-threatening, autosomal recessive genetic disorders characterized by acute crises of energy production and chronic energy deficiency. Patients may present with rhabdomyolysis induced by exercise; fasting or illness; hepatic dysfunction, including severe hypoglycemia and hyperammonemia; and cardiomyopathy. These clinical manifestations can lead to frequent hospitalizations and premature death. LC-FAODs are caused by mutations in nuclear genes encoding mitochondrial enzymes involved in the conversion of dietary long-chain fatty acids (LCFAs) into energy during times of fasting and physiologic stress. Despite newborn screening, current management options leave many patients continuing to experience major clinical events, and mortality rates remain elevated. The current standard therapy for LC-FAODs is avoidance of fasting and supplementation of medium-chain triglyceride oil, an even, medium-chain fatty acid that does not require the typical steps of LC-FAOD for metabolism. Despite this therapy, patients with LC-FAODs continue to experience recurring hospitalizations, and high morbidity and mortality rates. In recent years, the use of medium, odd-chain fatty acids, such as triheptanoin, have been studied as a treatment of LC-FAODs due to its anaplerotic properties. Due to favorable safety and efficacy data from clinical trials, this novel agent has the potential to transform the treatment of LC-FAODs and improve patient outcomes in this patient population. This article provides an overview of the epidemiology, pathophysiology, clinical manifestations, and current management approaches for the diagnosis and management of LC-FAODs. It also provides the most recent clinical safety and efficacy data for triheptanoin and other therapies under investigation.
Am J Manag Care. 2020;26:S147-S154. https://doi.org/10.37765/ajmc.2020.88480
Introduction
The Role of Fatty Acids in Production of Energy
Fatty acids provide a major energy source for the myocardium, skeletal muscle, and liver via β-oxidation, which occurs in the mitochondria.1 Short-chain and medium-chain fatty acids can enter the mitochondria directly, but long-chain fatty acids (LCFAs) must be transferred in via a shuttle involving 3 enzymes/transporters and carnitine.2 Once inside the mitochondrial matrix, β-oxidation progresses in a cycle of 4 steps, which result in the sequential cleavage of 2-carbon acetyl-coenzyme A (CoA) from the fatty acid chain and the transfer of electrons to the respiratory chain for adenosine triphosphate (ATP) production (Figure).2,3Acetyl-CoA can be utilized as a substrate in the tricarboxylic acid (TCA) cycle to create additional reducing equivalents for the electron transport chain, or it can be utilized for ketone synthesis, an alternate form of energy for the brain, myocardium, muscle, kidney, and other tissues.2 This energy is especially critical during periods of fasting when glucose is unavailable and during periods of physiologic stress. The metabolism of LCFAs to support creation of energy focuses on oxidation of acetyl-CoA in the mitochondrial TCA cycle.4 For proper TCA cycle function, inflow of substrates (anaplerosis) must be balanced by outflow (cataplerosis).5 In anaplerosis, intermediates are continuously replenished to sustain TCA cycle function and drive ATP production. With cataplerosis, other intermediates are removed to drive gluconeogenesis and lipogenesis.4,5 This balance is critical to maintaining energy homeostasis.4-6


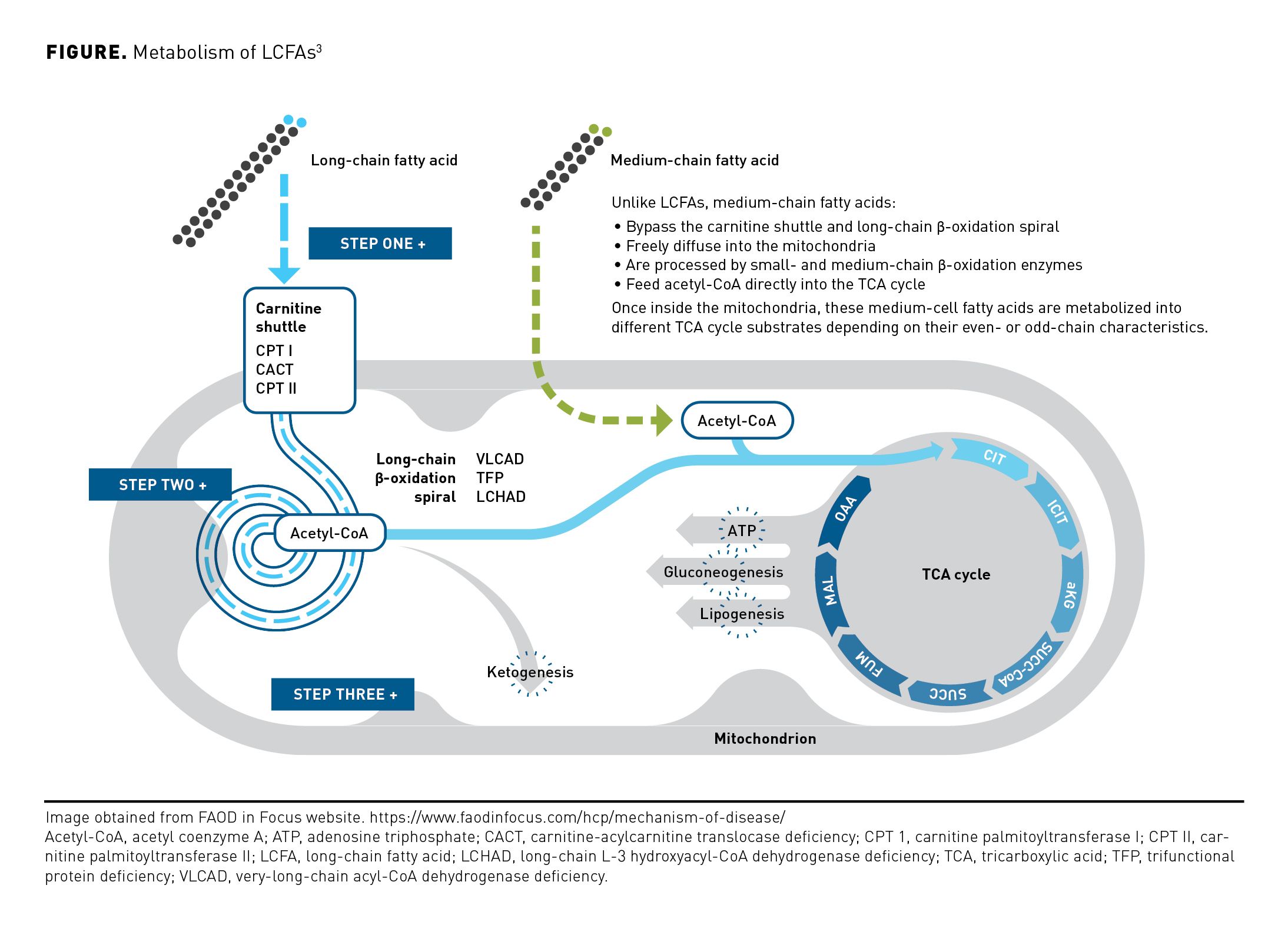
Fatty acids can be classified as even-chain or odd-chain based on the number of carbons in the α-carbon backbone. Fatty acids having an odd number of carbon atoms are minor physiologic species, with 16 and 18 carbon species predominating in food sources and tissue storage. Odd-chain fatty acids are oxidized identically to even-chain fatty acids, except that in the final round of β-oxidation, 1 molecule of each of acetyl-CoA and anaplerotic propionyl-Co are produced.7 The production of propionyl-CoA is specific to odd-carbon fatty acids and leads to their anaplerotic properties.8 Medium odd-chain fatty acids can restore energy production and improve cardiac and skeletal muscle function until odd-chain substrates become limiting, a problem eliminated by use of anaplerotic odd-chain fats.7,9
Overview of Long-Chain Fatty Acid Oxidation Disorders
Fatty acid oxidation disorders (FAODs) are a group of more than 20 life-threatening, inborn errors of metabolism caused by either disruption of entry of fatty acid substrates into mitochondria or a defect in their β-oxidation in the mitochondrial matrix.10,11 They are inherited as autosomal recessive conditions. Historically, they have been shown to lead to significant morbidity and mortality.9 Fatty acid oxidation (FAO) is characterized as long and medium, based on the α-carbon chain length of fatty acyl-CoA substrates. Long-chain fatty acid oxidation disorders (LC-FAODs) affect the metabolism of fats of greater than 8-12 carbons and are caused by deficiencies of 1 of 3 enzymes/carriers responsible for entry of long-chain fats into mitochondria (the carnitine cycle) or 4 steps of the mitochondrial β-oxidation spiral in the mitochondrial matrix. The end result is a deficit of reducing equivalents for mitochondrial oxidative phosphorylation. Symptoms in patients with LC-FAODs are driven by acute crises of energy production and chronic energy deficiency.8,12,13 In LC-FAODs, the oxidation of fatty acids is disrupted by deficiencies in crucial mitochondrial enzymes, which in turn compromises energy homeostasis and sparks a buildup of potentially toxic fatty acid intermediates.4 Partial or incomplete oxidation of fatty acids leads to accumulation of elevated concentrations of metabolite intermediates in blood and organs as well as systemic effects, such as hypoglycemia, hyperammonemia, and acidosis.14 Common symptoms include cardiomyopathy and recurrent rhabdomyolysis.
Incidence and Mortality Rates Associated With LC-FAODs
LC-FAODs occur with an overall incidence of 1 in 9300 individuals in the United States, Australia, and Germany, with a similar incidence in the rest of the world.15 In the United States and many developed countries, FAODs are identified by newborn screening (NBS). Annually, there are an estimated 100 births with a confirmed diagnosis in the United States, with an estimated 2000 to 3500 individuals currently living with an LC-FAOD.16 Before NBS, mortality was as high as 60% to 90%.14 In patients identified by NBS, mortality is lower, but morbidity remains high, with recurrent symptoms and a negative impact on quality of life.8,10
Incidence and Mortality Rates Associated With LC-FAODs
LC-FAODs occur with an overall incidence of 1 in 9300 individuals in the United States, Australia, and Germany, with a similar incidence in the rest of the world.15 In the United States and many developed countries, FAODs are identified by newborn screening (NBS). Annually, there are an estimated 100 births with a confirmed diagnosis in the United States, with an estimated 2000 to 3500 individuals currently living with an LC-FAOD.16 Before NBS, mortality was as high as 60% to 90%.14 In patients identified by NBS, mortality is lower, but morbidity remains high, with recurrent symptoms and a negative impact on quality of life.8,10
Clinical Symptoms of LC-FAODs
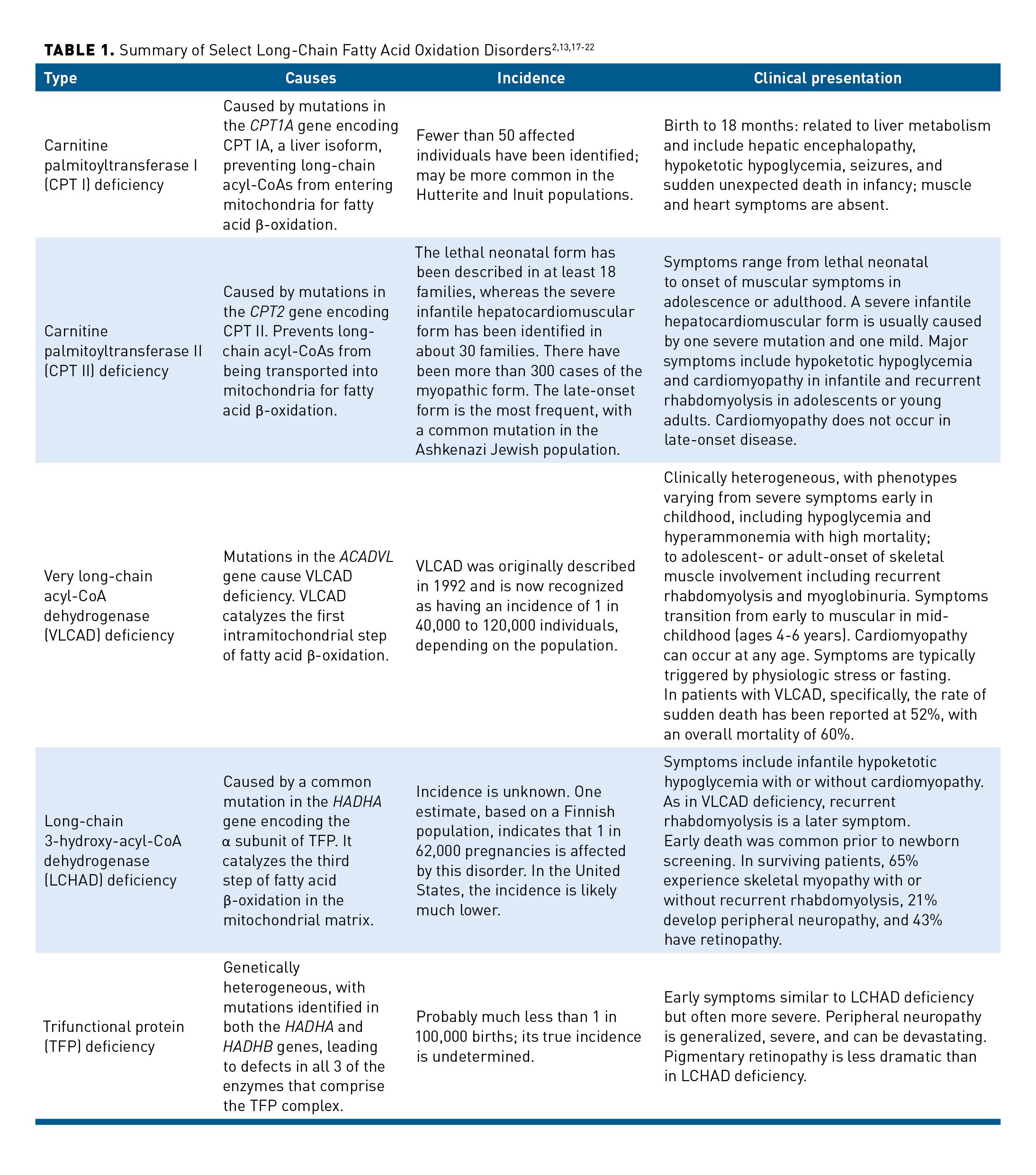
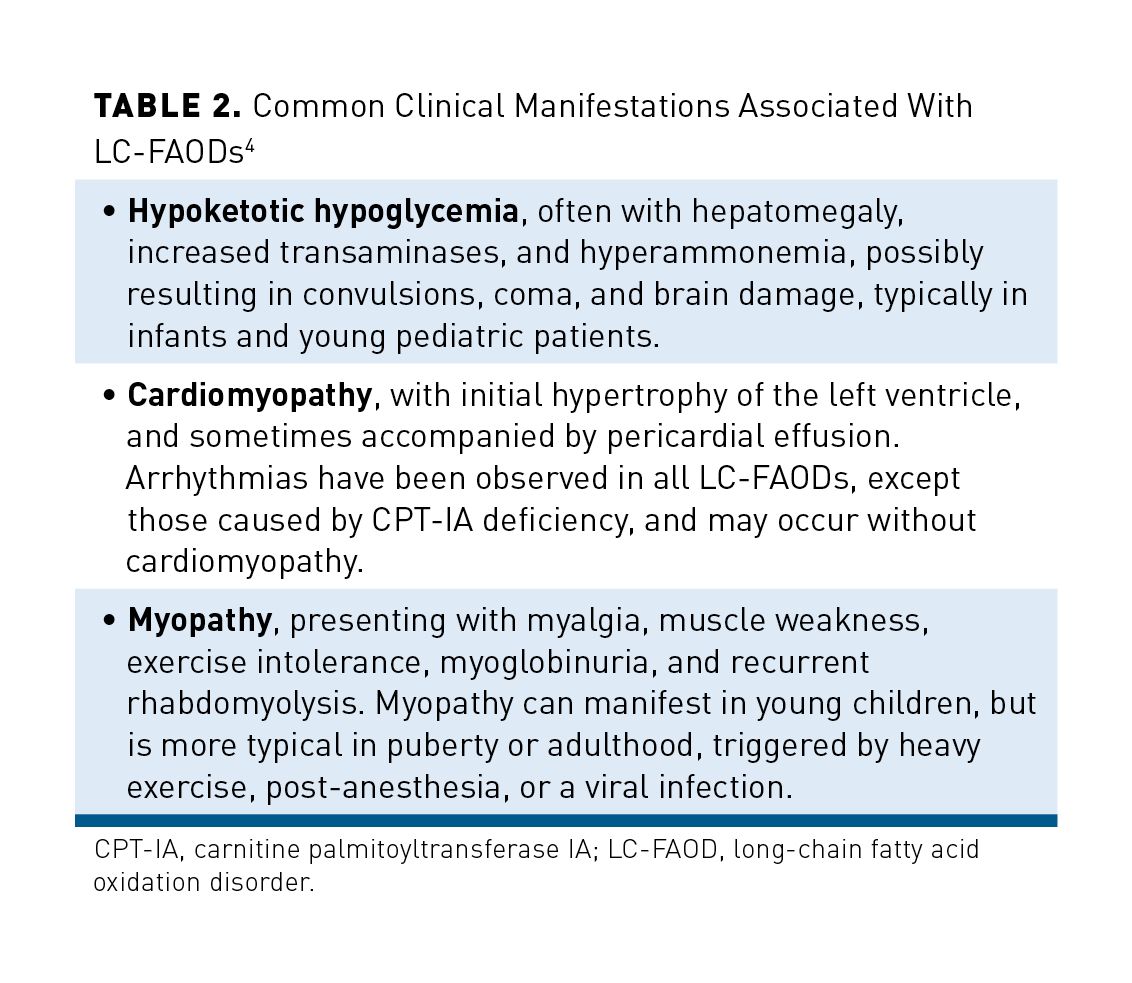
The defects of LC-FAODs include both the carnitine cycle and the intramitochondrial β-oxidation cycle. They include carnitine palmitoyltransferase (CPT I or CPT II) deficiency, very long-chain acyl-coA dehydrogenase (VLCAD) deficiency, long-chain 3-hydroxy-acyl-coA dehydrogenase (LCHAD) deficiency, and trifunctional protein (TFP) deficiency (Table 12,13,17-22). Of these, VLCAD deficiency is the most common.4 In general, clinical symptoms are more similar than different among the disorders and, as discussed above, include hypoglycemia, hyperammonemia, recurrent rhabdomyolysis, and acute or chronic cardiomyopathy (Table 24). Cardiomyopathy can be dilated or hypertrophic. The cardinal finding of hypoketotic hypoglycemia is variable and may not manifest in the milder forms of each disease. Hypoglycemia tends to dominate the clinical picture in infants and younger children, with rhabdomyolysis becoming more common in older children (>6 years of age). Cardiomyopathy can occur at any age. Peripheral neuropathy and retinal degeneration are unique to LCHAD/TFP deficiencies.10




NBS of LC-FAODs
Historically, LC-FAODs could only be diagnosed symptomatically, often after sudden or near death, as all clinical parameters returned to normal when patients were well. A retrospective analysis of 187 clinically diagnosed patients with LC-FAODs concluded that mortality rates had not changed overall during the 3 decades before the publication.14 Even with an increased understanding of pathophysiology and evolving standard of care, overall there was greater than 50% mortality in each decade in patients who received a diagnosis symptomatically. The development of acylcarnitine analysis via mass spectrometry allowed identification of many patients who were asymptomatic, but some level of clinical suspicion was necessary before the test was ordered. More recently, the United States and many developed nations have incorporated FAODs into expanded NBS using tandem mass spectrometry,13,23 allowing identification of affected infants before the development of symptoms.4 NBS and early intervention have diminished mortality rates for patients with FAODs; however, most continue to experience substantial morbidity due to episodes of metabolic decompensation despite treatment.4,9,10
Clinical Management of LC-FAODs
Even though the understanding of the pathophysiology of LC-FAODs has expanded considerably in the past few decades, treatments for LC-FAODs have largely involved nutritional and symptomatic management. Historically, treatment has been consensus based as evidence-driven outcomes are limited.13 In June 2020, an agent specifically indicated for the treatment of pediatric and adult patients with LC-FAOD, triheptanoin (Dojolvi), was approved and will be discussed below along with agents under investigation.24
The primary goal of nutrition management of LC-FAODs is to limit long-chain fat as a substrate for energy production, both by preventing β-oxidation and catabolism and by restricting the amount of dietary long-chain fat while still providing adequate nutrients for normal growth and development.10 The level to which fat is limited is reliant on the individual patient’s gene mutation and disease severity. Patients with LC-FAODs in mild or moderate forms typically experience intermittent symptoms at times of augmented β-oxidation, such as while exercising or fasting.10 Routine fasting overnight is safe for most patients, with a maximum of 8 to 10 hours recommended by most guidelines.10,25 Medium-chain triglyceride (MCT) oil given 30 to 45 minutes before strenuous exercise decreases accumulation of long-chain fats. Additional intake of carbohydrates and fluids should occur every 2 hours. Early implementation of sick-day protocol is recommended when symptoms of an intercurrent illness are present or with the development of rhabdomyolysis.25 Sick-day protocol can often be implemented at home with increased fluid and caloric intake to prevent catalysis, especially during febrile illnesses. However, if intake is inadequate or if myoglobinuria develops as detected with standard urinalysis dipsticks, then admission to the hospital is necessary. In the hospital, maintenance of caloric intake via an intravenous dextrose solution containing 10% dextrose with appropriate electrolytes at 1.5 times maintenance fluid infusion rate will provide 8 mg/kg/min of glucose, usually sufficient to suppress catabolism. Serum electrolytes, creatinine, and creatinine phosphokinase (CPK) should be monitored and treatment continued until the CPK is trending down. Decision to transition to enteral feeds and discharge from the hospital must be personalized.10,26
The use of carnitine supplementation in LC-FAODs has not been well studied and is controversial.13 Supplementation is often considered if secondary carnitine deficiency develops; however, some rodent studies have suggested that long-chain hydroxyl-acylcarnitines may induce arrhythmias.10 Patients require at least 10% of dietary calories from long-chain fats to maintain normal essential fatty acid levels and may need essential fatty acid supplementation if levels are low. Docosahexaenoic acid (DHA) is crucial for brain, visual, and immune functions and prevention of fat-soluble vitamin deficiencies and should be supplemented in LCHAD/TFP deficiency.10
Management in Infancy
Some age-specific management issues should be considered in LC-FAODs. Nutritional management in infancy should deliver 40% to 45% of total energy from fat, with a minimum of 10% from long-chain fat (often increased to 20% in mild disease).10,27 Breast milk is high in fat content and breastfeeding may need to be halted in symptomatic infants.10 Infants with mild or asymptomatic disease may be able to breastfeed, but alternating breastfeeding with metabolic formula containing MCTs or supplementing pumped breast milk with metabolic formula may be considered. These formulas are often prescribed by a metabolic physician/specialist.10
Childhood and Adult Management
In childhood and adulthood, reduction of calories from fat to 30% to 35% of total energy is appropriate. In severe disease, the patient may still need to limit long-chain fat to 10% of total energy and provide MCTs for the remainder of the energy requirement.10,27 However, a 10% long-chain fat diet is difficult to follow and relaxation to 20% is often allowed, similar to that of patients with milder disease. As formulas for infants with FAODs are enriched with essential fatty acids, essential fats may need to be supplemented when infant formula is halted.10 Linoleic acid (C18:2n6) and α-linolenic acid (C18:3n3) should comprise 3% and 1% of energy and energy intake, respectively, typically through supplementation with specific oils, such as walnut or flaxseed oil.10,27 Patients are at risk for becoming deficient in fat-soluble vitamins and may need supplementation. Additionally, fasting limitations of 8 to 10 hours are often employed.10,28
Proper Use of MCT Oils
Commercially available MCT oils consist of a mix of octanoate (C8:0), decanoate (C10:0), and some dodecanoate (C12:0) fatty acids esterified to a glycerol backbone with the proportions of these fatty acids varying from lot to lot.28 Prescription forms of MCT are essentially pure trioctanoylglycerol. In general, the daily dose of MCTs should be equally divided across all meals, sometimes with a dose reserved before excessive physical activity. Infants on metabolic formula typically tolerate MCT oil without symptoms, but older patients starting treatment for the first time experience gastrointestinal symptoms, including abdominal discomfort, cramping, flatulence, bloating, and diarrhea. In this instance, reducing the daily dose and increasing slowly to the desired final dose usually resolves symptoms. MCTs can be mixed into a variety of foods and beverages. Additional recommendations for using MCT oils are found in Table 3.29



Recently Approved Therapy
Triheptanoin
Following the discovery of LC-FAODs, it was generally assumed that supplementation for these patients with MCT oil would prevent clinical symptoms, as the even-chain fats it contained could completely bypass the metabolism. However, clinical symptoms persisted in patients, and is now recognized to be due at least in part to depletion of odd-chain TCA cycle intermediates and subsequent persistent energy deficiency. In recognition of this deficit, triheptanoin, a food-grade supplemental heptanoyl-triglyceride, was proposed as a more physiologically balanced MCT as 2 cycles of FAO produced both 2- and 3-carbon intermediates. Early case reports on efficacy led to a significant compassionate-use experience with the medication. Two retrospective studies demonstrated considerable improvement in treated patients, specifically reductions in episodes and hospital days related to hypoglycemia, cardiomyopathy, and rhabdomyolysis.12,30 This, in turn, led to the development and FDA approval of pharmaceutical-grade triheptanoin. Triheptanoin is available commercially as an oral liquid supplied in a 500-mL bottle (supplying 8.3 kcal/mL). The target daily dose is up to 35% of the patient’s total prescribed daily caloric intake divided into 4 or more doses administered at mealtimes or with snacks. Initial doses are 10% of the daily caloric intake and slowly increased to the target of 35% over 2 to 3 weeks.24 Upon ingestion, triheptanoin is rapidly digested to heptanoate in the small intestine and absorbed into the circulation. As with other medium-chain fats, heptanoate can diffuse across the mitochondrial membrane and be metabolized by the medium-chain–specific enzymes of FAO, bypassing the need for the carnitine cycle, the long-chain transport, and long-chain–specific enzymes.8 The 3-carbon propionyl-CoA product of FAO is further metabolized to succinyl-CoA and succinate, resupplying the TCA cycle intermediates that are secondarily deficient in patients and restoring the ATP production necessary for gluconeogenesis.8,31
Clinical Trials of Triheptanoin/UX007
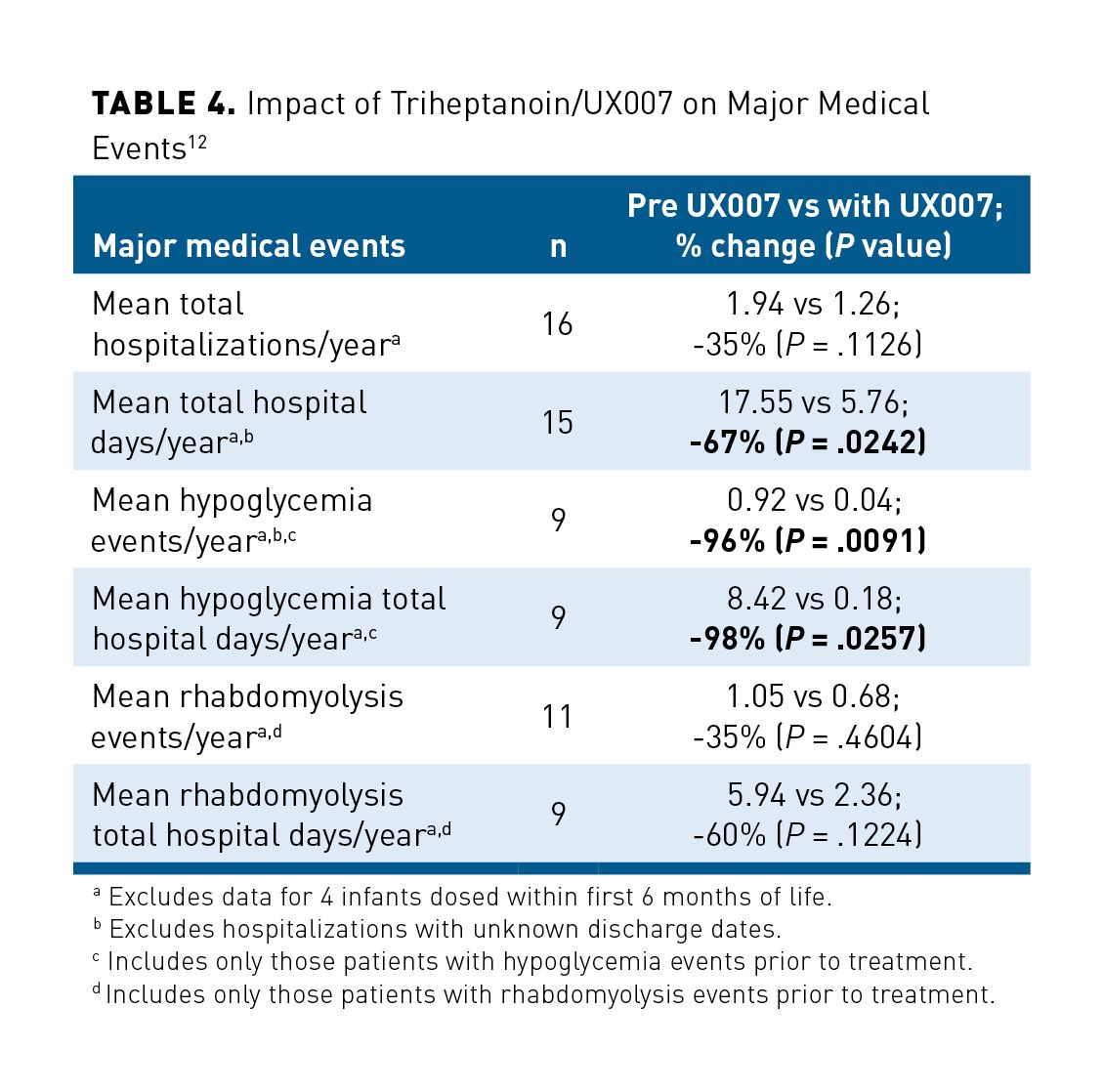
Clinical trials have assessed the safety and efficacy of triheptanoin in the treatment of LC-FAODs. The first was a double-blinded, randomized controlled study that compared changes in physiologic function after treatment with pharmaceutically pure octanoyl or heptanoylglycerol in a double-blind fashion in 32 participants. Patients in the C7 (triheptanoin) group experienced increased left ventricular (LV) ejection fraction by 7.4% (P = .046) while experiencing a 20% (P = .041) decrease in LV wall mass on their resting echocardiogram. They also required a lower heart rate (average = 6.98 beats per minute [bpm]) for the same amount of work during a moderate-intensity exercise stress test when compared with patients taking C8 (trioctanoin) (95% CI, 0.34-13.63 bpm; P = .040) There was no significant difference in total energy expenditure, phosphocreatine recovery with acute exercise, body composition, incidence of rhabdomyolysis, or any secondary outcome measures between the groups. There were no significant differences in adverse effects (AEs) between C7 and C8, and the majority were minor and gastrointestinal in nature. The improvement in cardiac function in hearts with no obvious dysfunction is in keeping with improved cardiac outcomes in other clinical trials, whereas the lack of a difference in clinical outcomes reflects the short duration of the study and the relative stability of patients entering the study.28 The second trial was a retrospective analysis of data from 20 of a total of 24 patients with LC-FAOD who were enrolled in a compassionate use protocol and treated with triheptanoin for up to 13 years. Hospitalization event rates, number of hospitalization days per year, and event rates for rhabdomyolysis, hypoglycemia, and cardiomyopathy were collected and compared before and after triheptanoin initiation (Table 412). A total of 320 hospitalizations were documented for 20 subjects from birth through the end of the study period. Mean hospitalization days per year decreased significantly by 67% after triheptanoin initiation (17.55 vs 5.76; P = .0242). Additionally, hypoglycemia events were nearly eliminated with a significant reduction in events per year and hospital days per year. Rhabdomyolysis events and hospitalization days trended down but were non-significantly changed with addition of triheptanoin. The data collected related to cardiomyopathy symptoms were insufficient to assess for impact of triheptanoin administration. AEs related to treatment with triheptanoin in the 20 patients included diarrhea (60%), gastrointestinal upset (55%), frequent loose stools (15%), and lethargy (10%).12


Results of a study for FDA registration of triheptanoin have also been published. This study was a single-arm, open-label, multicenter phase 2 safety and efficacy study evaluating patients with severe LC-FAOD evidenced by ongoing related musculoskeletal, cardiac, and/or hepatic events despite treatment; end points were at 24 and 78 weeks. Patients acted as their own control. After a 4-week run-in on current therapy, triheptanoin was titrated to a target dose of 25% to 35% of total daily caloric intake. The incidence of and hospitalizations related to hypoglycemia, cardiomyopathy, and rhabdomyolysis were captured and compared with the year prior to starting in the study. Patients also were evaluated on several age/condition-eligible end points, including submaximal exercise tests to assess muscle function/endurance 12-minute walk test (12MWT), exercise tolerance (cycle ergometry), and health-related quality of life (HRQOL). Twenty-nine patients (0.8-58 years) were enrolled; most qualified based on severe musculoskeletal disease. Twenty-five patients (86%) completed the 24-week treatment period. At week 18, eligible patients (n = 8) demonstrated a 28% increase (LS mean = +181.9 meters; P = .087) from baseline (673.4 meters) in 12MWT distance. At week 24, eligible patients (n = 7) showed a 60% increase in watts (W) generated (LS mean = +409.3W; P = .149) over baseline (744.6W) for the exercise tolerance test. Improvements in exercise tests were supported by significant improvements from baseline in the adult (n = 5) self-reported SF-12v2 physical component summary score (LS mean = +8.9; P <.001). In patients with severe LC-FAOD, triheptanoin interim study results revealed enhanced exercise endurance and tolerance and was linked with positive changes in self-reported HRQOL.9 At 78 weeks, there was a demonstrated persistent reduction of the incidence of major clinical events and related hospital days related to rhabdomyolysis, hypoglycemia, and cardiomyopathy. The composite end points of annualized event rate and annualized duration rate for all events were significantly reduced as noted in Table 5.8,13 The majority of AEs over both studies were mild to moderate gastrointestinal symptoms, including diarrhea (44%), vomiting (44%), nausea (14%), and abdominal pain (including abdominal discomfort, abdominal distention, upper abdominal pain, and gastrointestinal pain (60%). AEs led to dose reductions in 35% of patients in the first study and 12% of patients in the extension study. AEs were often managed with smaller, frequent doses mixed with food.8,24


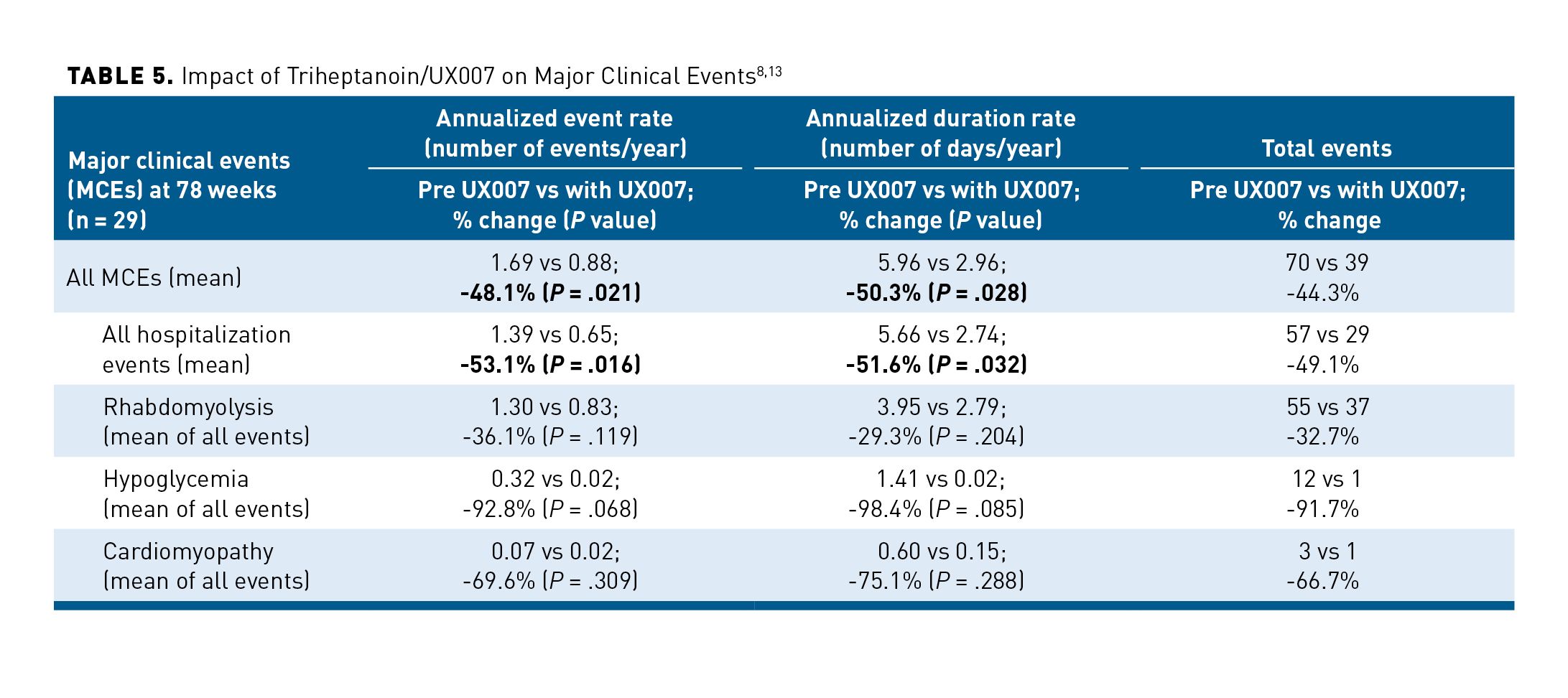
Subsequently, 75 patients from previous studies were enrolled in a long-term safety extension and efficacy open-label phase 2 study. Patients will begin or continue treatment with triheptanoin while maintaining other dietary restrictions. Primary outcome measures of this study include annualized LC-FAOD major clinical events (rhabdomyolysis, hypoglycemia, and cardiomyopathy) through 84 months on treatment and monitoring for number of treatment-emergent AEs or serious AEs. This study is underway with an estimated completion date of September 2021.32
Agents Under Development
The peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily, ligand-modulated transcription factors that regulate gene expression of many cellular processes. The 3 PPARs, α, γ, and δ, are activated by lipids and are targets for current drug therapies.33 PPARα is primarily expressed in the liver, where it upregulates genes involved in lipid oxidation in the fasted state.34-36 PPARγ is highly expressed in adipose tissue and regulates adipogenesis and insulin sensitivity.37 PPARδ controls genes involved in cellular metabolic processes, such as glucose homeostasis, fatty acid mobilization and oxidation, and electron transport chain. PPARδ is expressed in metabolically active tissues, including liver, muscle, and fat.38
Bezafibrate, a relatively broad-spectrum PPAR agonist that increases expressions of a number of FAO genes, has been investigated as a treatment for FAODs.25 Three clinical trials of bezafibrate have produced contradictory findings. The first trial, which was an open-label, 6-month pilot study with a 3-year extension, showed increased level of whole-body fat oxidation, improved QOL scores, reduced creatinine kinase levels, and diminished occurrence of rhabdomyolysis with bezafibrate.39,40 Results of a double-blind, randomized, placebo-controlled study failed to demonstrate a benefit of bezafibrate over placebo in clinical symptoms or FAO capacity during exercise.41 Finally, frequency of myopathic attacks was not reduced in an open-label study; however, QOL scores were improved.25,42 To date, there are no active trials for bezafibrate.
In September 2019, the FDA granted orphan drug designation to REN001, a selective PPARγ agonist, for the treatment of FAODs. REN001 is currently being investigated in a phase 1b trial involving 12 patients with confirmed FAODs. The primary outcome measure of the study is to assess the safety and tolerability of REN001.43
Conclusions
LC-FAODs are a heterogeneous group of disorders that have seen some improvement in clinical outcomes through early identification of patients with NBS but remain problematic, nonetheless. Traditional therapies still leave patients at risk for episodes of hypoglycemia, cardiomyopathy, and rhabdomyolysis common to all LC-FAODs, along with peripheral neuropathy and pigmentary retinopathy in LCHAD/TFP deficiencies. The recent FDA approval of triheptanoin is likely to improve—but not eliminate—these clinical problems. Available clinical studies demonstrate that triheptanoin directly addresses TCA cycle deficiencies that arise in LC-FAODs and improves clinical outcomes with reduction in clinical symptoms and hospital days. As such, triheptanoin represents a substantial breakthrough that will greatly improve the treatment and management of LC-FAODs. Thus, patients will continue to need careful monitoring by a team of experienced metabolic care providers working in close collaboration with patients’ primary care providers. Patients’ access to educational, financial, and support resources are also clear needs, given the long-term implications and the need for chronic access to the healthcare system.
Author affiliation: Jerry Vockley, MD, PhD, is Chief of the Division of Medical Genetics and Director of The Center for Rare Disease Therapy, UPMC Children’s Hospital of Pittsburgh; and Cleveland Family Endowed Chair in Pediatric Research and Professor of Human Genetics, University of Pittsburgh, both in Pittsburgh, PA.
Funding source: This activity is supported by an educational grant from Ultragenyx Pharmaceutical Inc.
Author disclosure: Dr Vockley has the following relevant financial relationships with commercial interests to disclose:
Grant/Research support – Reneo Pharmaceuticals, Ultragenyx Pharmaceutical Inc
Authorship information: Substantial contributions to the concept and design; analysis and interpretation of data; and critical revision of the manuscript for important intellectual content.
Address correspondence to: gerard.vockley@chp.edu
Medical writing and editorial support: Yvette C. Terrie, RPh, BS Pharm
REFERENCES
1. Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. 2016;1863(10):2422-2435. doi: 10.1016/j.bbamcr.2016.01.023
2. Sun A, Merritt JL II. Orphan drugs in development for long-chain fatty acid oxidation disorders: challenges and progress. Orphan Drugs: Res Rev. 2015;5:33-41. doi: 10.2147/ODRR.S63061
3. Healthy fatty acid oxidation. FAOD in Focus. Accessed July 10, 2020. faodinfocus.com/hcp/mechanism-of-disease
4. Knottnerus SJ, Bleeker JC, Wüst RC, et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord. 2018;19(1):93-106. doi: 10.1007/s11154-018-9448-1
5. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277(34):30409-30412. doi: 10.1074/jbc.R200006200
6. Brunengraber H, Roe CR. Anaplerotic molecules: current and future. J Inherit Metab Dis. 2006;29(2-3):327-331. doi: 10.1007/s10545-006-0320-1
7. Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110(2):259-269. doi: 10.1172/JCI15311
8. Vockley J, Burton B, Berry GT, et al. Results from a 78-week, single-arm, open-label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long-chain fatty acid oxidation disorders (LC-FAOD). J Inherit Metab Dis. 2019;42(1):169-177. doi: 10.1002/jimd.12038
9. Vockley J, Burton B, Berry GT, et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: safety and efficacy in children and adults following 24 weeks of treatment. Mol Genet Metab. 2017;120(4):370-377. doi: 10.1016/j.ymgme.2017.02.005
10. Merritt JL II, Norris M, Kanungo S. Fatty acid oxidation disorders. Ann Transl Med. 2018;6(24):473. doi: 10.21037/atm.2018.10.57
11. Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57(5 Pt 2):78R-86R. doi: 10.1203/01.PDR.0000159631.63843.3E
12. Vockley J, Marsden D, McCracken E, et al. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment—a retrospective chart review [published correction appears in Mol Genet Metab. 2015;116(3):221]. Mol Genet Metab. 2015;116(1-2):53-60. doi: 10.1016/j.ymgme.2015.06.006
13. Spiekerkoetter U, Lindner M, Santer R, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32(4):488-497. doi: 10.1007/s10545-009-1125-9
14. Baruteau J, Sachs P, Broue P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013;36(5):795-803. doi: 10.1007/s10545-012-9542-6
15. Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33(5):521-526. doi: 10.1007/s10545-010-9076-8
16. Diagnosing LC-FAOD. FAOD in Focus. 2020. Accessed July 10, 2020. faodinfocus.com/diagnosis
17. Taggart RT, Smail D, Apolito C, Vladutiu GD. Novel mutations associated with carnitine palmitoyltransferase II deficiency. Hum Mutat. 1999;13(3):210-220. doi: 10.1002/(SICI)1098-1004(1999)13:3<210::AID-HUMU5>3.0.CO;2-0
18. Carnitine palmitoyltransferase I deficiency. Genetics Home Reference. Reviewed April 2014. Published June 9, 2020. Accessed June 22, 2020. ghr.nlm.nih.gov/condition/carnitine-palmitoyltransferase-i-deficiency#
19. Carnitine palmitoyltransferase II deficiency. Genetics Home Reference. Reviewed June 2014. Published June 9, 2020. Accessed June 22, 2020. ghr.nlm.nih.gov/condition/carnitine-palmitoyltransferase-ii-deficiency#genes
20. Very long-chain acyl-CoA dehydrogenase deficiency. Genetics Home Reference. Reviewed February 2018. Published June 9, 2020. Accessed June 22, 2020. ghr.nlm.nih.gov/condition/very-long-chain-acyl-coa-dehydrogenase-deficiency#definition
21. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Genetics Home Reference. Reviewed May 2017. Published June 9, 2020. Accessed June 2020. ghr.nlm.nih.gov/condition/long-chain-3-hydroxyacyl-coa-dehydrogenase-deficiency#statistics
22. Mitochondrial trifunctional protein deficiency. Genetics Home Reference. Reviewed September 2019. Published June 9, 2020. Accessed June 22, 2020. ghr.nlm.nih.gov/condition/mitochondrial-trifunctional-protein-deficiency#genes
23. Rocha H, Castiñeiras D, Delgado C, et al. Birth prevalence of fatty acid β-oxidation disorders in Iberia. JIMD Rep. 2014;16:89-94. doi: 10.1007/8904_2014_324
24. Dojolvi. Prescribing information. Ultragenyx Pharmaceutical Inc; 2020. Accessed July 15, 2020. ultragenyx.com/medicines/dojolvi-full-prescribing-information/
25. Yamada K, Taketani T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet. 2019;64(2):73-85. doi: 10.1038/s10038-018-0527-7
26. Aldubayan SH, Rodan LH, Berry GT, Levy HL. Acute illness protocol for fatty acid oxidation and carnitine disorders. Pediatr Emerg Care. 2017;33(4):296-301. doi: 10.1097/PEC.0000000000001093
27. Rohr F, van Calcar S. Very long chain acyl CoA dehydrogenase deficiency (VLCADD). Genetic Metabolic Dietitians International: Nutrition Guidelines. Updated September 4, 2008. Accessed May 20, 2020. gmdi.org/Resources/Nutrition-Guidelines/VLCAD
28. Gillingham MB, Heitner SB, Martin J, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40(6):831-843. doi: 10.1007/s10545-017-0085-8
29. Shah ND, Limketkai BN. The use of medium-chain triglycerides in gastrointestinal disorders. Practical Gastro. Published February 2017. Accessed May 20, 2020. med.virginia.edu/ginutrition/wp-content/uploads/sites/199/2014/06/Parrish-February-17.pdf
30. Vockley J, Charrow J, Ganesh J, et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016;119(3):223-231. doi: 10.1016/j.ymgme.2016.08.008
31. Marin-Valencia I, Good LB, Ma Q, Malloy CR, Pascual JM. Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J Cereb Blood Flow Metab. 2013;33(2):175-182. doi: 10.1038/jcbfm.2012.151
32. Long-Chain Fatty Acid Oxidation Disorders (LC-FAOD) Extension Study for Subjects Previously Enrolled in Triheptanoin Studies. Updated June 4, 2020. Accessed July 10, 2020. clinicaltrials.gov/ct2/show/NCT02214160
33. Hansen MK, Connolly TM. Nuclear receptors as drug targets in obesity, dyslipidemia and atherosclerosis. Curr Opin Investig Drugs. 2008;9(3):247-255.
34. Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A. 1997;94(9):4312-4317. doi: 10.1073/pnas.94.9.4312
35. Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1993;103(11):1489-1498. doi: 10.1172/JCI6223
36. Xu HE, Lambert MH, Montana VG, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3(3):397-403. doi: 10.1016/s1097-2765(00)80467-0
37. Hevener AL, He W, Barak Y, et al. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003;9(12):1491-1497. doi: 10.1038/nm956
38. Oliver WR Jr, Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98(9):5306-5311. doi: 10.1073/pnas.091021198
39. Bonnefont JP, Bastin J, Behin A, et al. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360(8):838-840. doi: 10.1056/NEJMc0806334
40. Bonnefont JP, Bastin J, Laforet P, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. 2010;88(1):101-108. doi: 10.1038/clpt.2010.5
41. Ørngreen MC, Madsen KL, Preisler N, Andersen G, Vissing J, Laforêt P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. 2014;82(7):607-613. doi: 10.1212/WNL.0000000000000118
42. Shiraishi H, Yamada K, Oki E, et al. Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan; 2nd report QOL survey. Mol Genet Metab Rep. 2019;20:100496. doi: 10.1016/j.ymgmr.2019.100496
43. A Study of the Safety of REN001 in Patients With Fatty Acid Oxidation Disorders. Updated August 26, 2019. Accessed July 15, 2020. clinicaltrials.gov/ct2/show/NCT03833128
Click here to download PDF version
