
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Identification, Course, and Management of Progressive Pulmonary Fibrosis

ABSTRACT
The term “progressive pulmonary fibrosis” or “PPF” is generally used to describe progressive lung fibrosis in an individual with an interstitial lung disease (ILD) other than idiopathic pulmonary fibrosis (IPF). Several sets of criteria have been proposed for the identification of PPF, most of which are based on a combination of a decline in forced vital capacity, worsening of respiratory symptoms, and increase in the extent of fibrosis on radiology. Although some risk factors for faster progression of fibrosing ILD have been identified, it remains challenging to predict which individuals will develop PPF. Close monitoring, including regular pulmonary function tests, is required to detect the earliest signs of worsening disease. PPF is associated with high rates of hospitalization and death. Management of PPF requires a multidisciplinary and multimodal approach, including pharmacological therapy and supportive care. Discussions about palliative care should begin at an early stage, individualized to the needs of the patient.
Am J Manag Care. 2024;30:S122-S130. https://doi.org/10.37765/ajmc.2024.89634
For author information and disclosures, see end of text.



Introduction
Interstitial lung diseases (ILDs) comprise a large group of disorders characterized by inflammation and/or fibrosis of the lung parenchyma. Some ILDs have a known cause, for example, ILDs related to autoimmune diseases1 or occupational exposures,2 while others are idiopathic, the most common of which is idiopathic pulmonary fibrosis (IPF).3 Environmental factors, such as exposures to dusts, molds, or chemicals,4 and genetic variants5 contribute to the development of ILD. A proportion of patients with ILDs develops pulmonary fibrosis that becomes progressive. The term “progressive pulmonary fibrosis” (PPF) or “progressive fibrosing ILD” (PF-ILD) is generally used to describe progressive lung fibrosis in an individual with an ILD other than IPF (which is, by definition, a progressive disease).3 This article describes the identification, course, and management of PPF.
Criteria for Defining PPF
Several sets of criteria have been proposed for the identification of PPF, mainly based on expert consensus and extrapolation from what is known about disease behavior and prognosis in IPF.3,6-10 The most widely used criteria are those developed to identify patients with PPF for enrollment in the INBUILD trial of nintedanib6 and those later proposed in a clinical practice guideline published by the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Asociacion Latinoamericana de Torax (ATS/ERS/JRS/ALAT) in May 2022 (Table).3,6 Most definitions of PPF include some combination of a decline in forced vital capacity (FVC) and/or diffusing capacity for carbon monoxide (DLCO), a worsening of respiratory symptoms, and an increase in the extent of fibrosis on high-resolution CT (HRCT). However, there is variability in terms of the magnitude of change in these parameters that should define progression and the time period over which progression should be assessed. The ATS/ERS/JRS/ALAT guideline stated that the criteria for identification of PPF may be fulfilled at any time within a 1-year period,3 while other criteria use a 2-year window.6,7,9,10 It is important to note that progression may occur at any time within the specified time period and needs not be assessed over its whole duration. Further, some experts have argued that no time frame should be applied when assessing progression in clinical practice, as progression is predictive of a poor prognosis regardless of the time period over which it occurs.11



In addition to the debate over the criteria that should be used to define PPF, discussion continues over whether PPF should be identified only in patients in whom standard management for the respective ILD has failed. A requirement for patients to have progressed despite standard management was included in the inclusion criteria for the INBUILD trial of nintedanib in patients with PPF,6 the RELIEF trial of pirfenidone in patients with PPF,9 and a trial of pirfenidone in patients with progressive fibrotic hypersensitivity pneumonitis.12 However, in many fibrotic ILDs, “standard management” is not well defined, and varies based on patient characteristics, drug availability and coverage, and other factors. The ATS/ERS/JRS/ALAT guideline did not mandate that patients receive standard management for ILD prior to PPF being identified; however, its conditional recommendation for the use of nintedanib in patients with PPF was limited to patients who have failed standard management.3 A consensus statement published by an expert group in 2023 suggested that a distinction should be made between a first presentation of PPF and PPF that has occurred despite management of the ILD, as this may have discrete treatment implications.13
Risk Factors for Development of PPF
PPF affects patients with a wide range of ILDs.14-17 Different studies have provided broad estimates for the risk of PPF in patients with ILDs, depending on the population, criteria, and methodology.18-21 Among 1749 patients with non-IPF fibrotic ILDs receiving care at 9 ILD referral centers, 14% met the INBUILD trial criteria for PPF.22 In a study of registry data from 753 patients with non-IPF fibrotic ILDs, 30% of patients met the international guideline criteria for PPF, 37% met the INBUILD trial criteria, 32% met the criteria from the trial of pirfenidone in patients with unclassifiable ILD, and 23% met the RELIEF trial criteria.21 In an analysis of claims data, 30,771 patients were identified as having a fibrosing ILD other than IPF and, of these, 47% were predicted to have progressive disease.18 Children with fibrosing ILDs such as those related to genetic diseases, autoimmune diseases, or exposures may also develop progressive lung fibrosis,23 but the proportions of pediatric patients who fulfill specific criteria for PPF are unknown.
Different types of ILD appear to be associated with differing risks of PPF. In a retrospective analysis of 1227 consecutive patients with non-IPF fibrotic ILDs, the proportion of patients with PPF (defined as a relative decline in FVC % predicted ≥ 10%, or a relative decline in FVC % predicted ≥ 5 to < 10% plus any 1 of the following: hospitalization, increased extent of fibrosis on HRCT, increased oxygen use, or respiratory death, within 2 years) ranged from 10.3% in patients with ILD due to sarcoidosis to 41.9% in patients with unclassifiable idiopathic interstitial pneumonia.21
Several risk factors for progression of fibrotic ILDs have been identified. Demographic risk factors for progression include older age24,25 and male sex.26,27 Cigarette smoking is also a risk factor.26,28 A lower FVC15,29 or diffusing capacity of the lung for carbon monoxide (DLCO)30,31 is also associated with a greater risk of short-term progression. Among 2746 patients in the CARE-PF registry, FVC < 70% predicted and DLCO < 75% predicted at baseline were independent risk factors for the development of PPF (defined as a relative decline in FVC % predicted ≥ 10%, death, lung transplant, or any 2 of a relative decline in FVC % predicted between ≥ 5 and < 10%, worsening respiratory symptoms, or worsening fibrosis on HRCT) over 2 years.25 On radiology, a greater extent of lung fibrosis15,24,32,33 and a usual interstitial pneumonia (UIP) pattern24,26,34,35 are associated with faster progression. Genetic risk factors for faster progression of fibrotic ILDs have also been identified, such as mutations associated with short telomeres.36
Although some risk factors have been identified, it remains challenging to predict which individuals with fibrosing ILD will progress, when, and at what rate. Recognition of PPF requires close monitoring of disease behavior to detect the earliest clinical, physiologic, and/ or radiologic signs of worsening disease. No biomarker has been shown to have sufficient accuracy to predict progression before it occurs. Patients with fibrosing ILDs should be monitored with regular symptom assessment and pulmonary function tests (PFTs) and repeat HRCT as clinically indicated, so that if progression has occurred, it is detected promptly.13,37-39 Experts in the management of patients with ILD suggest that PFTs should be performed at least every 3 to 4 months.13,38 HRCT should be performed less frequently, but may be valuable for assessing progression in patients in whom lung function has declined or symptoms have worsened.13,37,39 Repeated HRCT may be appropriate in some patients (eg, every 12 to 24 months to evaluate for complications), as an adjunct to PFTs, to assess whether patients meet PPF criteria, as well as to screen for lung cancer,40 which may be more common in patients with ILD.41 Other evaluations, such as a 6-minute walk test, may also provide valuable information about the progression of a patient’s ILD.42
Course of PPF
The pathobiology of fibrosis involves fibroproliferation and excess production of extracellular matrix, leading to destruction of the lung architecture.43,44 The mechanisms that drive fibrosis are selfperpetuating43,44 and, in the absence of treatment, PPF will inevitably progress. Even in patients receiving proven therapies for PPF, lung function may continue to decline.45-50 There is some evidence that decline in FVC may be more rapid in patients with PPF who have a UIP-like fibrotic pattern on HRCT.51-53 Heterogeneity in the trajectory of FVC decline has been observed among patients with PPF and differing ILD diagnoses. In a retrospective analysis, the decline in FVC in patients with PPF due to autoimmune diseases was significantly smaller than in patients with PPF and either hypersensitivity pneumonitis or idiopathic interstitial pneumonia.53 Differing risks of mortality have also been observed among patients with PPF and different ILD diagnoses (Figure 1).18,52


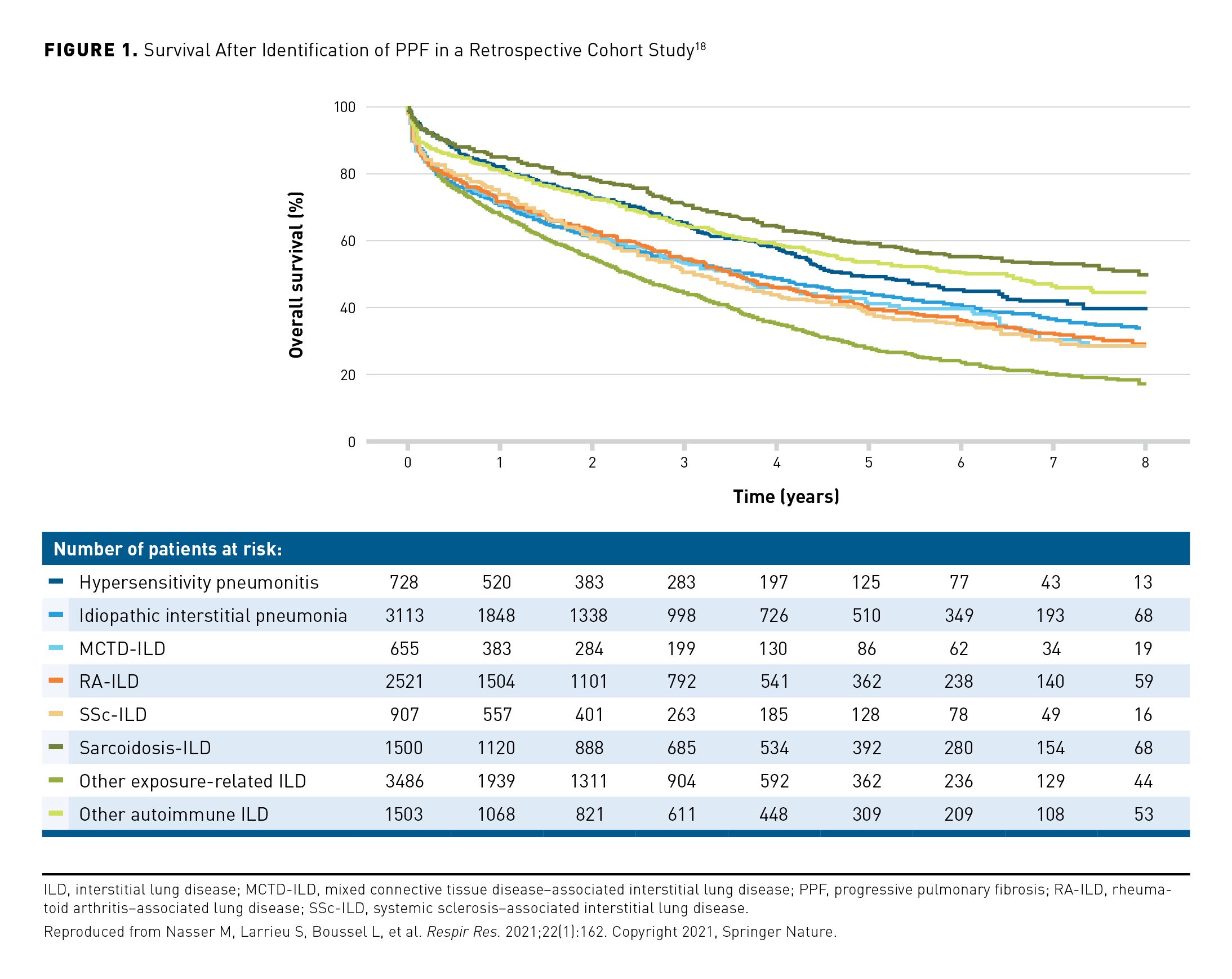
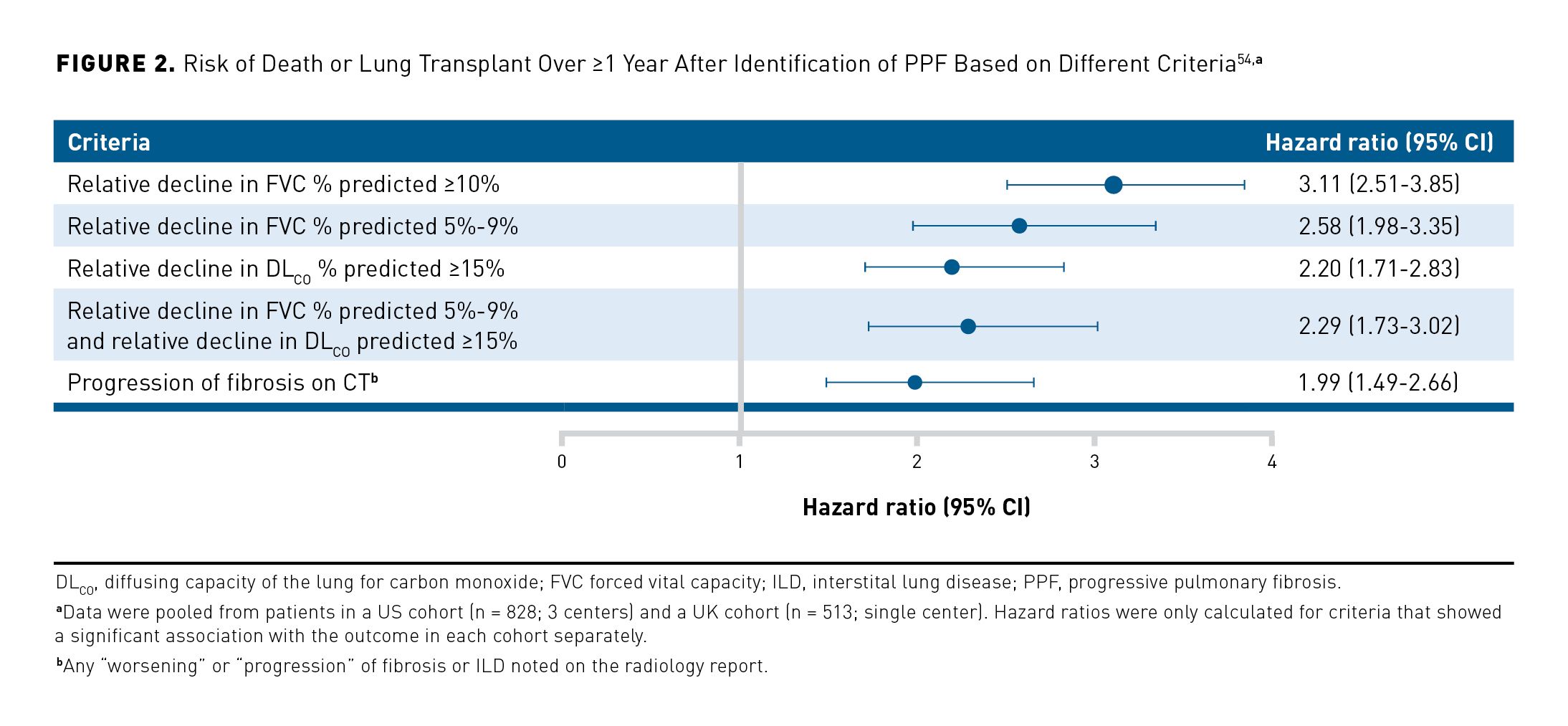
Patients identified as having PPF using different criteria show differences in short-term mortality.22,25,54 In a retrospective analysis of 1341 patients, the risk of death or lung transplant over ≥ 1 year was greater in patients who had PPF defined as a decline in FVC % predicted of > 10% than in patients who met other criteria indicating disease progression (Figure 2).54 However, it is important to bear in mind that regardless of how PPF is identified, it is associated with high rates of hospitalization and death.18,19,22,25,54-56 In a descriptive analysis, over a mean follow-up of 69 months, 46% of 135 patients with PPF had a respiratory hospitalization compared with 22% of 261 patients with fibrosing ILD but without PPF at the same center.55 In the same study, 5-year survival was 64.5% in the patients with PPF compared with 89.5% in the patients without PPF.55 Patients with non-IPF fibrosing ILDs may experience acute lung function deterioration (acute exacerbation), which is associated with high mortality.57-60


Monitoring of PPF
PPF progresses at a variable rate, which is not necessarily linear in an individual patient.53,61 Further, lung function decline may not correlate with a patient’s perception of their health status or symptoms.62,63 It is important that patients with PPF are regularly monitored so that progression can be evaluated and management, including the provision of supportive/palliative care, adjusted if necessary. Monitoring should comprise PFTs, assessment of symptoms, and where clinically indicated, repeat imaging. As well as monitoring disease progression, any adverse effects (AEs) of therapies should be identified and managed; the potential of immunosuppressant and antifibrotic therapies to cause AEs should not be underestimated.
Management of PPF
The management of PPF requires that patients receive an accurate diagnosis of the underlying ILD, ideally in the context of a multidisciplinary discussion.64 This has implications for prognosis and guides the optimal approach to management. Many ILDs have an identifiable cause or associated condition, which may require distinct treatment approaches. The work-up includes obtaining a detailed medical, family, occupational, environmental, and social history and conducting a physical examination. Tests include PFTs, HRCT scans, and serologic testing to identify potential exposures and exclude autoimmune diseases. For patients who have undergone bronchoscopy or surgical biopsies, histopathological features should be integrated with clinical findings in multidisciplinary discussion, including, at minimum, input from pulmonology, radiology, and pathology.65-68 In patients with hypersensitivity pneumonitis, identification and avoidance of the inciting antigen is associated with improved survival28 and may help to prevent progression.69
Management of PPF requires a multidisciplinary and multimodal approach (Figure 3).37,38,69 Patients should be given adequate information about their disease and emotional support.70-72 Supportive care focuses on symptom management and preserving quality of life and may include pulmonary rehabilitation, which can improve exercise capacity, quality of life and dyspnea.73 Oxygen use is recommended for patients with ILD and severe chronic resting hypoxemia or severe exertional hypoxemia.74 Worsening of health status, such as decline in physical functioning or initiation of oxygen, should prompt a consideration of palliative care to help manage symptoms and quality of life, individualized to the patient’s physical, emotional, and spiritual needs.71 Advance care planning, and discussion of the patient’s goals and preferences for end-of-life care, should be initiated as early as possible.75,76 Referral for lung transplantation should be considered in individuals with PPF who meet relevant criteria.77



Pharmacological Therapy for PPF
Pharmacologic therapy for PPF differs based on the ILD diagnosis. The standard treatment for ILD associated with autoimmune disease is immunosuppression, with antifibrotic therapy in certain cases. Randomized controlled trials have shown that cyclophosphamide,78 mycophenolate,79 nintedanib,80 tocilizumab,81 and rituximab82 slow the progression of ILD associated with systemic sclerosis (SSc-ILD). Nintedanib and tocilizumab have been approved by the FDA for the treatment of SSc-ILD. A clinical practice guideline for the treatment of SSc-ILD published by the ATS in October 2023 included a strong recommendation for the use of mycophenolate and conditional recommendations for the use of cyclophosphamide, tocilizumab, nintedanib, nintedanib plus mycophenolate, and rituximab.83 A guideline for the treatment of ILD in patients with autoimmune disease issued by the American College of Rheumatology (ACR) and American College of Chest Physicians (CHEST)84 provided similar recommendations for the treatment of SSc-ILD and conditional recommendations for first-line treatment of autoimmune ILDs other than SSc-ILD for mycophenolate, azathioprine, rituximab, and cyclophosphamide.
Immunosuppressants are commonly used in patients with hypersensitivity pneumonitis,85,86 and have shown benefits in some patients, but it is not clear whether they slow the progression of fibrotic forms of the disease, which often behave similarly to IPF.87-90 The use of corticosteroids alone or combined with immunomodulatory therapy is not recommended in patients with IPF91,92 and combination therapy with prednisone, azathioprine, and N-acetylcysteine is harmful in these patients.93 Immunosuppressant therapies, including rituximab and tocilizumab, are associated with an increased risk of infection.82,90,94 In addition, mycophenolate and azathioprine are associated with gastrointestinal AEs79,86,88 and cyclophosphamide with leukopenia and neutropenia.78
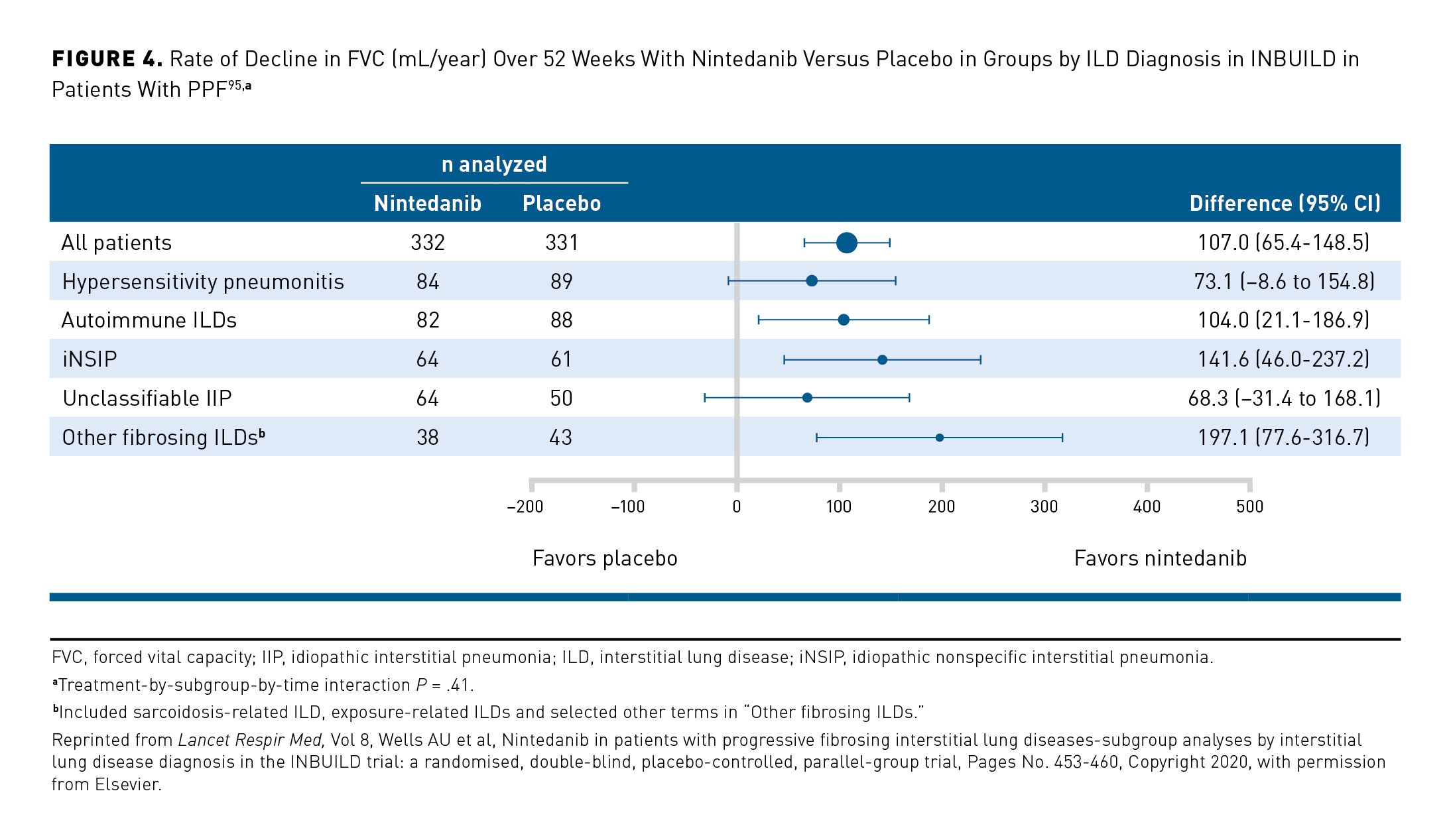
In patients with PPF, clinicians should consider use of a treatment shown to slow its progression. In the US, the FDA has approved nintedanib for the treatment of progressive fibrosing ILDs of any etiology. ATS/ERS/JRS/ALAT guidelines provide a conditional recommendation for nintedanib as a treatment option for patients with PPF for whom standard management (not defined) has failed.3 In the INBUILD trial in 663 patients with PPF despite management in clinical practice, nintedanib slowed the rate of decline in FVC over 52 weeks by 57% compared with placebo (–81 mL/year in the nintedanib group compared with –188 mL/year in the placebo group; P <.001),6 with a consistent effect among patients with different diagnoses (Figure 4)95 and disease severities96 and evidence of a significant benefit on respiratory symptoms based on patients’ responses to the Living with Pulmonary Fibrosis (L-PF) questionnaire.97 The AEs associated with nintedanib are mainly gastrointestinal, particularly diarrhea, which was reported in 66.9% of patients in the nintedanib group and 23.9% of patients in the placebo group over 52 weeks of the INBUILD trial.6 Pirfenidone, an approved treatment for IPF, has also been investigated in randomized placebo-controlled trials in patients with other progressive fibrosing ILDs. In a trial in patients with progressive unclassifiable fibrosing ILD, the decline in FVC (mL) over 24 weeks was lower in the pirfenidone group (n = 118) (–17.8 mL) than in the placebo group (n = 119) (–113.0 mL) (P = .002).8 In the TRAIL1 trial in 123 patients with rheumatoid arthritis–associated ILD, pirfenidone slowed the decline in FVC over 52 weeks versus placebo (–66 vs –146 mL/year; P = .0082).98 In the RELIEF trial, among 67 patients with progressive fibrosing ILDs related to autoimmune diseases, nonspecific interstitial pneumonia, hypersensitivity pneumonitis, or asbestosis, the decline in FVC over 48 weeks was numerically lower in patients treated with pirfenidone than placebo (–36.6 mL vs –114.4 mL), but the difference was not statistically significant (P = .21).9 Pirfenidone is not licensed in any country for the treatment of ILDs other than IPF. The ATS/ERS/JRS/ALAT guidelines issued a recommendation for more research into the efficacy of pirfenidone in PPF.3 AEs of pirfenidone include gastrointestinal discomfort and photosensitivity. In a meta-analysis of data from patients with PPF, pirfenidone use was associated with an increased risk of gastrointestinal discomfort (relative risk, 1.83; 95% CI, 1.29-2.60) and photosensitivity (relative risk, 4.88; 95% CI, 1.09-21.83) versus placebo.99


Comorbidities such as gastroesophageal reflux disease, obstructive sleep apnea, pulmonary hypertension, and heart failure are common in patients with PPF,55,56,100 and should be screened for and managed, as they can contribute to symptoms and morbidity. Pulmonary hypertension is associated with reduced exercise tolerance and poor prognosis in patients with ILDs.101-103 In the INCREASE study in patients with pulmonary hypertension (documented by right heart catheterization) due to ILD, inhaled treprostinil was associated with an improvement in 6-minute walk test distance compared with placebo at week 16 (primary end point [mean difference vs placebo], 31.12 m; 95% CI, 16.85-45.39; P <.001).104 Based on these results, the FDA approved inhaled treprostinil to improve exercise capacity in patients with pulmonary hypertension associated with ILD.
The randomized, placebo-controlled InPedILD trial investigated the effects of nintedanib, administered using weight-based dosing, in 39 children and adolescents with fibrosing ILD.47 The proportion of patients with AEs over 24 weeks (primary end point relating to safety) was 84.6% in both the nintedanib and placebo groups. Diarrhea, the most frequent AE, was reported in 38.5% of patients in the nintedanib group and 15.4% of patients in the placebo group.47 The trial was not powered to show a difference in FVC, but changes in FVC % predicted over 24 weeks were numerically in favor of nintedanib.47
Conclusions
While various definitions are used for PPF, most include a combination of decline in lung function, worsening of symptoms, and increase in radiological abnormalities. In most trials, PPF was identified only in patients who had failed “standard management” for their particular ILD, but in most ILDs, standard management is not well defined. Patients with worse lung function, a greater extent of fibrosis on HRCT, or a UIP pattern on HRCT are at higher risk of progression of their ILD. However, predicting the onset of PPF remains challenging. PPF confers a poor prognosis and needs to be detected promptly. It is important that patients with fibrosing ILDs are closely monitored, including with regular lung function tests. Management of PPF requires a multimodal approach, including pharmacological therapy, supportive care, and the identification and treatment of comorbidities. Advance care planning and palliative care should be discussed with the patient from an early stage.
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors. Editorial support was provided by Elizabeth Ng and Wendy Morris of Fleishman-Hillard, London, United Kingdom, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Author Affiliations: Division of Pulmonary, Critical Care, and Sleep Medicine, National Jewish Health (EFP), Denver, CO; Department of Medicine, Division of Pulmonary and Critical Care, Weill Cornell Medicine (AJP), New York, NY.
Source of Funding: This article is part of a supplement that was supported by Boehringer Ingelheim Pharmaceuticals, Inc. The authors did not receive any payment for their work on the articles. Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Author Disclosures: Dr Podolanczuk reports participating in consultancies or paid advisory boards for Boehringer Ingelheim, United Therapeutics, and Veracyte. Dr Fernandez Perez reports no relationship or financial interest with any entity that would pose a conflict of interest with the subject matter of this supplement.
Authorship Information: Concept and design (AJP, EFP); analysis and interpretation of data (AJP); drafting of the manuscript (AJP, EFP); critical revision of the manuscript for important intellectual content (AJP, EFP); and supervision (AJP).
Address Correspondence to: Anna J. Podolanczuk, MD. Weill Cornell Medicine, 425 East 61st St, New York, NY, 10065. Email: ajp9012@med.cornell.edu
REFERENCES
1. Panagopoulos P, Goules A, Hoffmann-Vold AM, Matteson EL, Tzioufas A. Natural history and screening of interstitial lung disease in systemic autoimmune rheumatic disorders. Ther Adv Musculoskelet Dis. 2021;13:1759720X211037519. doi:10.1177/1759720X211037519
2. Blanc PD, Annesi-Maesano I, Balmes JR, et al. The occupational burden of nonmalignant respiratory diseases. An official American Thoracic Society and European Respiratory Society statement. Am J Respir Crit Care Med. 2019;199(11):1312-1334. doi:10.1164/rccm.201904-0717ST
3. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47. doi:10.1164/rccm.202202-0399ST
4. Copeland CR, Collins BF, Salisbury ML. Identification and remediation of environmental exposures in patients with interstitial lung disease: evidence review and practical considerations. Chest. 2021;160(1):219-230. doi:10.1016/j.chest.2021.02.021
5. Borie R, Le Guen P, Ghanem M, Taillé C, et al. The genetics of interstitial lung diseases. Eur Respir Rev. 2019;28(153):190053. doi:10.1183/16000617.0053-2019
6. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718-1727. doi:10.1056/NEJMoa1908681
7. George PM, Spagnolo P, Kreuter M, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. 2020;8(9):925-934. doi:10.1016/S2213-2600(20)30355-6
8. Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8(2):147-157. doi:10.1016/S2213-2600(19)30341-8
9. Behr J, Prasse A, Kreuter M, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. 2021;9(5):476-486. doi:10.1016/S2213-2600(20)30554-3
10. Lobo LJ, Liu Y, Li P, et al. Characteristics of patients with progressive fibrosing interstitial lung diseases (ILDs) in the ILD-PRO Registry. Abstract presented at the 2023 Annual Meeting of the American College of Chest Physicians; October 8-11, 2023; Honolulu, HI. Available at: https://www.usscicomms.com/respiratory/CHEST2023/Lobo2
11. Cottin V, Brown KK, Flaherty KR, Wells AU. Progressive pulmonary fibrosis: should the timelines be taken out of the definition? Am J Respir Crit Care Med. 2022;206(10):1293-1294. doi:10.1164/rccm.202206-1143LE
12. Fernández Pérez ER, Crooks JL, Lynch DA, et al. Pirfenidone in fibrotic hypersensitivity pneumonitis: a double-blind, randomised clinical trial of efficacy and safety. Thorax. 2023;78(11):1097-1104. doi:10.1136/thorax-2022-219795
13. Rajan SK, Cottin V, Dhar R, et al. Progressive pulmonary fibrosis: an expert group consensus statement. Eur Respir J. 2023;61(3):2103187. doi:10.1183/13993003.03187-2021
14. Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc. 2018;15(4):460-469. doi:10.1513/AnnalsATS.201704-288OC
15. Hoffmann-Vold AM, Fretheim H, Halse AK, et al. Tracking impact of interstitial lung disease in systemic sclerosis in a complete nationwide cohort. Am J Respir Crit Care Med. 2019;200(10):1258-1266. doi:10.1164/rccm.201903-0486OC
16. Huang S, Doyle TJ, Hammer MM, et al. Rheumatoid arthritis-related lung disease detected on clinical chest computed tomography imaging: prevalence, risk factors, and impact on mortality. Semin Arthritis Rheum. 2020;50(6):1216-1225. doi:10.1016/j.semarthrit.2020.08.015
17. Lee JK, Ahn Y, Noh HN, et al. Clinical effect of progressive pulmonary fibrosis on patients with connective tissue disease-associated interstitial lung disease: a single center retrospective cohort study. Clin Exp Med. 2023;23(8):4797-4807. doi:10.1007/s10238-023-01212-z
18. Nasser M, Larrieu S, Boussel L, et al. Estimates of epidemiology, mortality and disease burden associated with progressive fibrosing interstitial lung disease in France (the PROGRESS study). Respir Res. 2021;22(1):162. doi:10.1186/s12931-021-01749-1
19. Singer D, Bengtson LGS, Conoscenti CS, et al. Claims-based prevalence of disease progression among patients with fibrosing interstitial lung disease other than idiopathic pulmonary fibrosis in the United States. Ann Am Thorac Soc. 2022;19(7):1112-1121. doi:10.1513/AnnalsATS.202102-222OC
20. Hilberg O, Hoffmann-Vold AM, Smith V, et al. Epidemiology of interstitial lung diseases and their progressive-fibrosing behaviour in six European countries. ERJ Open Res. 2022;8(1):00597-2021. doi:10.1183/23120541.00597-2021
21. Khor YH, Farooqi M, Hambly N, et al. Patient characteristics and survival for progressive pulmonary fibrosis using different definitions. Am J Respir Crit Care Med. 2023;207(1):102-105. doi:10.1164/rccm.202205-0910LE
22. Simpson T, Barratt SL, Beirne P, et al. The burden of progressive fibrotic interstitial lung disease across the UK. Eur Respir J. 2021;58(1):2100221. doi:10.1183/13993003.00221-2021
23. Nevel RJ, Deutsch GH, Craven D, et al. The US national registry for childhood interstitial and diffuse lung disease: report of study design and initial enrollment cohort. Pediatr Pulmonol. 2024;59(9):2236-2246. doi:10.1002/ppul.26568
24. Kim HC, Lee JS, Lee EY, et al. Risk prediction model in rheumatoid arthritis-associated interstitial lung disease. Respirology. 2020;25(12):1257-1264. doi:10.1111/resp.13848
25. Hambly N, Farooqi MM, Dvorkin-Gheva A, et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J. 2022;60(4):2102571. doi:10.1183/13993003.02571-2021
26. Chan C, Ryerson CJ, Dunne JV, Wilcox PG. Demographic and clinical predictors of progression and mortality in connective tissue disease-associated interstitial lung disease: a retrospective cohort study. BMC Pulm Med. 2019;19(1):192. doi:10.1186/s12890-019-0943-2
27. Ghuman A, Khanna D, Lin CJF, et al. Prognostic and predictive markers of systemic sclerosis-interstitial lung disease in a clinical trial and long-term observational cohort. Rheumatology (Oxford). 2024;63(2):472-481. doi:10.1093/rheumatology/kead234
28. Fernández Pérez ER, Swigris JJ, Forssén AV, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644-1651. doi:10.1378/chest.12-2685
29. Mooney JJ, Elicker BM, Urbania TH, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144(2):586-592. doi:10.1378/chest.12-2623
30. Hyldgaard C, Ellingsen T, Hilberg O, Bendstrup E. Rheumatoid arthritis-associated interstitial lung disease: clinical characteristics and predictors of mortality. Respiration. 2019;98(5):455-460. doi:10.1159/000502551
31. Ojanguren I, Morell F, Ramón MA, et al. Long-term outcomes in chronic hypersensitivity pneumonitis. Allergy. 2019;74(5):944-952. doi:10.1111/all.13692
32. Jeny F, Uzunhan Y, Lacroix M, et al. Predictors of mortality in fibrosing pulmonary sarcoidosis. Respir Med. 2020;169:105997. doi:10.1016/j.rmed.2020.105997
33. Chen X, Guo J, Yu D, Jie B, Zhou Y. Predictors of mortality in progressive fibrosing interstitial lung diseases. Front Pharmacol. 2021;12:754851. doi:10.3389/fphar.2021.754851
34. Adegunsoye A, Oldham JM, Bellam SK, et al. Computed tomography honeycombing identifies a progressive fibrotic phenotype with increased mortality across diverse interstitial lung diseases. Ann Am Thorac Soc. 2019;16(5):580-588. doi:10.1513/AnnalsATS.201807-443OC
35. Salisbury ML, Gu T, Murray S, et al. Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest. 2019;155(4):699-711. doi:10.1016/j.chest.2018.08.1076
36. Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710-1720. doi:10.1183/13993003.00308-2016
37. Nambiar AM, Walker CM, Sparks JA. Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians. Ther Adv Respir Dis. 2021;15:17534666211039771. doi:10.1177/17534666211039771
38. Bendstrup E, Kronborg-White S, Møller J, Prior TS. Current best clinical practices for monitoring of interstitial lung disease. Expert Rev Respir Med. 2022;16(11-12):1153-1166. doi:10.1080/17476348.2022.2162504
39. Koduri G, Solomon JJ. Identification, monitoring, and management of rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. 2023;75(12):2067-2077. doi:10.1002/art.42640
40. MacMahon H, Naidich DP, Goo JM, et al. Guidelines for management of incidental pulmonary nodules detected on CT images: from the Fleischner Society 2017. Radiology. 2017;284(1):228-243. doi:10.1148/radiol.2017161659
41. Naccache JM, Gibiot Q, Monnet I, et al. Lung cancer and interstitial lung disease: a literature review. J Thorac Dis. 2018;10(6):3829-3844. doi:10.21037/jtd.2018.05.75
42. Khor YH, Farooqi M, Hambly N, et al. Trajectories and prognostic significance of 6-minute walk test parameters in fibrotic interstitial lung disease: a multicenter study. Chest. 2023;163(2):345-357. doi:10.1016/j.chest.2022.08.2233
43. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol. 2019;15(12):705-730. doi:10.1038/s41584-019-0322-7
44. Rackow AR, Nagel DJ, McCarthy C, et al. The self-fulfilling prophecy of pulmonary fibrosis: a selective inspection of pathological signalling loops. Eur Respir J. 2020;56(5):2000075. doi:10.1183/13993003.00075-2020
45. Crestani B, Huggins JT, Kaye M, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med. 2019;7(1):60-68. doi:10.1016/S2213-2600(18)30339-4
46. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. Eur Respir J. 2022;59(3):2004538. doi:10.1183/13993003.04538-2020
47. Deterding R, Young LR, DeBoer EM, et al. Nintedanib in children and adolescents with fibrosing interstitial lung diseases. Eur Respir J. 2023;61(2):2201512. doi:10.1183/13993003.01512-2022
48. Raman L, Stewart I, Barratt SL, et al. Nintedanib for non-IPF progressive pulmonary fibrosis: 12-month outcome data from a real-world multicentre observational study. ERJ Open Res. 2023;9(2):00423-2022. doi:10.1183/23120541.00423-2022
49. Wuyts WA, Bonella F, Chaudhuri N, et al. Continued nintedanib treatment in patients with progressive pulmonary fibrosis: data from INBUILD-ON. Poster presented at: the European Respiratory Society (ERS) International Congress 2023; September 9-13, 2023; Milan, Italy. Available at: https://www.globalmedcomms.com/respiratory/ERS2023/Wuyts1
50. Allanore Y, Vonk MC, Distler O, et al. Continued treatment with nintedanib in patients with systemic sclerosis-associated interstitial lung disease: data from SENSCIS-ON. Ann Rheum Dis. 2022;81(12):1722-1729. doi:10.1136/ard-2022-222564
51. Brown KK, Inoue Y, Flaherty KR, et al. Predictors of mortality in subjects with progressive fibrosing interstitial lung diseases. Respirology. 2022;27(4):294-300. doi:10.1111/resp.14231
52. Nasser M, Larrieu S, Si-Mohamed S, et al. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J. 2021;57(2):2002718. doi:10.1183/13993003.02718-2020
53. Oldham JM, Lee CT, Wu Z, et al. Lung function trajectory in progressive fibrosing interstitial lung disease. Eur Respir J. 2022;59(6):2101396. doi:10.1183/13993003.01396-2021
54. Pugashetti JV, Adegunsoye A, Wu Z, et al. Validation of proposed criteria for progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2023;207(1):69-76. doi:10.1164/rccm.202201-0124OC
55. Kwon BS, Choe J, Chae EJ, Hwang HS, Kim YG, Song JW. Progressive fibrosing interstitial lung disease: prevalence and clinical outcome. Respir Res. 2021;22(1):282. doi:10.1186/s12931-021-01879-6
56. Singer D, Chastek B, Sargent A, et al. Impact of chronic fibrosing interstitial lung disease on healthcare use: association between fvc decline and inpatient hospitalization. BMC Pulm Med. 2023;23(1):337. doi:10.1186/s12890-023-02637-8
57. Kang J, Kim YJ, Choe J, Chae EJ, Song JW. Acute exacerbation of fibrotic hypersensitivity pneumonitis: incidence and outcomes. Respir Res. 2021;22(1):152. doi:10.1186/s12931-021-01748-2
58. Enomoto N, Naoi H, Mochizuka Y, et al. Frequency, proportion of PF-ILD, and prognostic factors in patients with acute exacerbation of ILD related to systemic autoimmune diseases. BMC Pulm Med. 2022;22(1):387. doi:10.1186/s12890-022-02197-3
59. Kwon BS, Lee HY, Choe J, Chae EJ, Hong S, Song JW. Acute respiratory deterioration in rheumatoid arthritis-associated interstitial lung disease: a single-center study. Chest. 2022;162(1):136-144. doi:10.1016/j.chest.2022.01.007
60. Tsubouchi K, Hamada N, Tokunaga S, et al Survival and acute exacerbation for patients with idiopathic pulmonary fibrosis (IPF) or non-IPF idiopathic interstitial pneumonias: 5-year follow-up analysis of a prospective multi-institutional patient registry. BMJ Open Respir Res. 2023;10(1):e001864. doi:10.1136/bmjresp-2023-001864
61. Maher TM, Brown KK, Kreuter M, et al. Effects of nintedanib by inclusion criteria for progression of interstitial lung disease. Eur Respir J. 2022;59(2):2004587. doi:10.1183/13993003.04587-2020
62. Swigris JJ, Wilson H, Esser D, et al. Psychometric properties of the St George’s Respiratory Questionnaire in patients with idiopathic pulmonary fibrosis: insights from the INPULSIS trials. BMJ Open Respir Res. 2018;18;5(1):e000278. doi:10.1136/bmjresp-2018-000278
63. Birring SS, Bushnell DM, Baldwin M, et al. The psychometric properties of the King’s Brief Interstitial Lung Disease questionnaire and thresholds for meaningful treatment response in patients with progressive fibrosing interstitial lung diseases. Eur Respir J. 2022;59(6):2101790. doi:10.1183/13993003.01790-2021
64. Graney BA, He C, Marll M, et al. Essential components of an interstitial lung disease clinic: results from a Delphi survey and patient focus group analysis. Chest. 2021;159(4):1517-1530. doi:10.1016/j.chest.2020.09.256
65. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med. 2018;6(2):138-153. doi:10.1016/S2213-2600(17)30433-2
66. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
67. Raghu G, Remy-Jardin M, Ryerson CJ, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2020;202(3):e36-e69. doi:10.1164/rccm.202005-2032ST
68. Fernández Pérez ER, Travis WD, Lynch DA, et al. Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest. 2021;160(2):e97-e156. doi:10.1016/j.chest.2021.03.066
69. Fernández Pérez ER, Koelsch TL, Leone PM, Groshong SD, Lynch DA, Brown KK. Clinical decision-making in hypersensitivity pneumonitis: diagnosis and management. Semin Respir Crit Care Med. 2020;41(2):214-228. doi:10.1055/s-0040-1701250
70. Oliveira A, Fabbri G, Gille T, et al. Holistic management of patients with progressive pulmonary fibrosis. Breathe (Sheff). 2023;19(3):230101. doi:10.1183/20734735.0101-2023
71. Palmer E, Kavanagh E, Visram S, Bourke AM, Forrest I, Exley C. When should palliative care be introduced for people with progressive fibrotic interstitial lung disease? A meta-ethnography of the experiences of people with end-stage interstitial lung disease and their family carers. Palliat Med. 2022;36(8):1171-1185. doi:10.1177/02692163221101753
72. Vick B, Vick P, Hoy H. Living with lung fibrosis: knowledge is power. Future Rare Dis. 2023;3(4). doi:10.2217/frd-2023-0003
73. Rochester CL, Alison JA, Carlin B, et al. Pulmonary rehabilitation for adults with chronic respiratory disease: an official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2023;208(4):e7-e26. doi:10.1164/rccm.202306-1066ST
74. Jacobs SS, Krishnan JA, Lederer DJ, et al. Home oxygen therapy for adults with chronic lung disease. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2020;202(10):e121-e141. doi:10.1164/rccm.202009-3608ST
75. Hjorth NE, Haugen DF, Schaufel MA. Advance care planning in life-threatening pulmonary disease: a focus group study. ERJ Open Res. 2018;4(2):00101-2017. doi:10.1183/23120541.00101-2017
76. Kalluri M, Orenstein S, Archibald N, Pooler C. Advance care planning needs in idiopathic pulmonary fibrosis: a qualitative study. Am J Hosp Palliat Care. 2022;39(6):641-651. doi:10.1177/10499091211041724
77. Leard LE, Holm AM, Valapour M, et al. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379. doi:10.1016/j.healun.2021.07.005
78. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655-2666. Doi:10.1056/NEJMoa055120
79. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708-719. doi:10.1016/S2213-2600(16)30152-7
80. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380(26):2518-2528. doi:10.1056/NEJMoa1903076
81. Roofeh D, Lin CJF, Goldin J, et al. Tocilizumab prevents progression of early systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. 2021;73(7):1301-1310. doi:10.1002/art.41668
82. Ebata S, Yoshizaki A, Oba K, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol. 2021;3(7):E489-E497. doi.org/10.1016/S2665-9913(21)00107-7
83. Raghu G, Montesi SB, Silver RM, et al. Treatment of systemic sclerosis-associated interstitial lung disease: evidence-based recommendations. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2024;209(2):137-152. doi:10.1164/rccm.202306-1113ST
84. Johnson SR, Bernstein EJ, Bolster MB, et al. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) guideline for the treatment of interstitial lung disease in people with systemic autoimmune rheumatic diseases. Arthritis Rheumatol. 2024;76(8):1182-1200. doi:10.1002/art.42861
85. Wijsenbeek M, Kreuter M, Olson A, et al. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35(11):2015-2024. doi:10.1080/03007995.2019.1647040
86. Wong AW, Khor YH, Donohoe K, et al. Prescribing patterns and tolerability of mycophenolate and azathioprine in patients with nonidiopathic pulmonary fibrosis fibrotic interstitial lung disease. Ann Am Thorac Soc. 2022;19(5):863-867. doi:10.1513/AnnalsATS.202108-954RL
87. Adegunsoye A, Oldham JM, Fernández Pérez ER, et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res. 2017;3(3):00016-2017. doi:10.1183/23120541.00016-2017
88. Morisset J, Johannson KA, Vittinghoff E, et al. Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest. 2017;151(3):619-625. doi:10.1016/j.chest.2016.10.029
89. De Sadeleer LJ, Hermans F, De Dycker E, et al. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. J Clin Med. 2018;8(1):14. doi:10.3390/jcm8010014
90. Ferreira M, Borie R, Crestani B, et al. Efficacy and safety of rituximab in patients with chronic hypersensitivity pneumonitis (cHP): a retrospective, multicentric, observational study. Respir Med. 2020;172:106146. doi:10.1016/j.rmed.2020.106146
91. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824. doi:10.1164/rccm.2009-040GL
92. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3-19. doi:10.1164/rccm.201506-1063ST
93. Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968-1977. doi:10.1056/NEJMoa1113354
94. Khanna D, Lin CJF, Furst DE, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020;8(10):963-974. doi:10.1016/S2213-2600(20)30318-0
95. Wells AU, Flaherty KR, Brown KK, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 2020;8(5):453-460. doi:10.1016/S2213-2600(20)30036-9
96. Kolb M, Flaherty KR, Silva RS, et al. Effect of nintedanib in patients with progressive pulmonary fibrosis in subgroups with differing baseline characteristics. Adv Ther. 2023;40(12):5536-5546. doi:10.1007/s12325-023-02668-x
97. Wijsenbeek M, Swigris JJ, Inoue Y, et al. Effects of nintedanib on symptoms in patients with progressive pulmonary fibrosis. Eur Respir J. 2024;63(2):2300752. doi:10.1183/13993003.00752-2023
98. Solomon JJ, Danoff SK, Woodhead FA, et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir Med. 2023;11(1):87-96. doi:10.1016/S2213-2600(22)00260-0
99. Ghazipura M, Mammen MJ, Bissell BD, et al. Pirfenidone in progressive pulmonary fibrosis: a systematic review and meta-analysis. Ann Am Thorac Soc. 2022;19(6):1030-1039. doi:10.1513/AnnalsATS.202103-342OC
100. Wong AW, Lee TY, Johannson KA, et al. A cluster-based analysis evaluating the impact of comorbidities in fibrotic interstitial lung disease. Respir Res. 2020;21(1):322. doi:10.1186/s12931-020-01579-7
101. Andersen CU, Mellemkjær S, Hilberg O, Nielsen-Kudsk JE, Simonsen U, Bendstrup E. Pulmonary hypertension in interstitial lung disease: prevalence, prognosis and 6 min walk test. Respir Med. 2012;106(6):875-882. doi:10.1016/j.rmed.2012.02.015
102. Hoeper MM, Behr J, Held M, et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015;10(12):e0141911. doi:10.1371/journal.pone.0141911
103. Alhamad EH, Cal JG, Alrajhi NN, Alharbi WM. Predictors of mortality in patients with interstitial lung disease-associated pulmonary hypertension. J Clin Med. 2020;9(12):3828. doi:10.3390/jcm9123828
104. Waxman A, Restrepo-Jaramillo R, Thenappan T, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325-334. doi:10.1056/NEJMoa2008470

