
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Hypertrophic Cardiomyopathy Considerations for the Managed Care Pharmacist
Abstract
Hypertrophic cardiomyopathy (HCM) is often seen in patients as an autosomal dominant genetic heart disease with a variable clinical course. It is characterized by left ventricular hypertrophy, and with some patients, there is no evidence of a genetic etiology or presence of HCM in family members. Young age at diagnosis and the presence of a pathogenic or likely pathogenic sarcomere variant predict greater lifelong risk for stroke, heart failure, ventricular arrhythmia, atrial fibrillation, or mortality. Most individuals affected with HCM live to an average lifespan due to improvements in earlier diagnosis, sudden cardiac death risk stratification, family screening, pharmacologic therapy, devices, and invasive septal reduction therapy. Although these interventions have improved mortality, they are associated with significant costs and morbidities. There are burdensome costs related to genetic testing, family screening, implantable cardioverter-defibrillators, alcohol septal ablation, septal myectomy, pacemaker placement, and cardiac transplantation. In addition to these economic considerations, patients with HCM may experience a diminished health-related quality of life. Shared decision making between the patient and physician, use of multidisciplinary teams at HCM centers, and judicious use of exercise when appropriate have been shown to improve patient outcomes. Currently, treatments for HCM do not treat the underlying illness. Although not yet approved by the FDA, cardiac myosin inhibitors have recently shown promise in clinical trials to treat the underlying pathology of HCM. If approved by the FDA, managed care pharmacists should be ready to assess their safety and efficacy to improve the clinical burden and quality of life of those affected by HCM and reduce medical costs for these patients against standard of care. Long-term safety and efficacy data showing reductions in hospitalizations, morbidity, and mortality will be needed to determine their actual utility in managing HCM and ultimate place in therapy.
Am J Manag Care. 2021;27(suppl 6):S118-S125. https://doi.org/10.37765/ajmc.2021.88629
Introduction
Hypertrophic cardiomyopathy (HCM) is often seen in patients as an autosomal dominant genetic heart disease that equally affects men and women with a heterogeneous clinical course. However, women are not diagnosed as frequently as men. The prevalence of asymptomatic HCM in young adults is approximately 1:200 to 1:500 in the United States. In adults, the incidence of symptomatic HCM based on medical claims data is less than 1:3000 adults in the United States. However, the real burden is much greater when accounting for undiagnosed disease in the general population. Assessment of HCM may be precipitated by the appearance of symptoms, an abnormal electrocardiogram during a routine physical examination, a cardiac event, detection of a heart murmur, or cardiac imaging during family screening studies.1
HCM is a disorder characterized by left ventricular hypertrophy (LVH) not attributable to other cardiac, systemic, or metabolic causes with or without genetic evidence of a disease-causing sarcomere variant. A maximal end-diastolic wall thickness of 15 mm or greater anywhere in the left ventricle verified by imaging (cardiovascular magnetic resonance or 2D echocardiography) or 13-14 mm along with the presence of HCM in a family member or positive genetic test in the absence of other causes confirms a clinical diagnosis of HCM in adults.1
Approximately 30% to 60% of patients with HCM have a detectable pathogenic or likely pathogenic genetic variant. Variants in one of eight or more genes encoding cardiac sarcomere proteins or sarcomere-related structures cause the LVH of HCM. The 2 most common pathogenic sarcomeric gene variants are myosin-binding protein C3 (MYBPC3) and beta myosin heavy chain 7 (MYH7), present in 70% of variant-positive patients. Other genes (ACTC1, MYL2, MYL3, TNNI3, TNNT2, TPM1) each account for about 1% to 5% of patients. Although family members with a pathogenic variant will likely develop clinical HCM, there is variability of age of disease occurrence.1
Clinical Burden
Although some patients with HCM may have an average life expectancy without debilitating symptoms or need for devices/surgery, other patients may experience a more severe disease course. Patients with a pathogenic sarcomeric gene variant or those diagnosed with HCM early in life may be at a greater lifelong risk for stroke, heart failure (HF), ventricular arrhythmia, atrial fibrillation (AF), or mortality. Approximately 30% to 40 % of patients with HCM may experience HF symptoms associated with systolic dysfunction, AF with increased risk for thromboembolic stroke, progressive symptoms due to diastolic dysfunction or left ventricular outflow tract obstruction (LVOTO), or sudden cardiac death (SCD). Thirty percent of patients develop nonobstructive HCM (nHCM), while most patients develop obstructive HCM (oHCM) over time. HCM mortality rates have decreased to less than 1% per year with modern therapies, such as implantable cardiac defibrillators (ICDs) and alcohol septal ablation (ASA), and SCD risk-stratification strategies. Due to the decrease in SCD rates, HF is now the predominant cause of morbidity and mortality in patients with HCM.1
A study involving the international Sarcomeric Human Cardiomyopathy Registry provided additional insights into factors related to poorer outcomes. Patients with HCM aged 20 to 29 years and 50 to 69 years had a greater than 4 times and equal to or greater than 3 times mortality, respectively, than the general US population. A 77% (95% CI, 0.72-0.80) cumulative incidence of the overall composite outcome by 60 years occurred in patients younger than 40 years at diagnosis, compared with 32% (95% CI, 0.29-0.36) by age 70 years for patients diagnosed older than 60 years. The overall composite outcome was defined as the first occurrence of SCD, cardiac arrest, ICD implantation, cardiac transplantation (CT), left ventricular assist device (LVAD) implantation, New York Heart Association (NYHA) functional class III/IV symptoms, AF, stroke, and death. A pathogenic or likely pathogenic sarcomere genetic variant predicted greater than 2-fold increased risk for all outcomes, with the chance for ventricular arrhythmias being the highest (HR, 2.8; 95% CI, 2.1-3.9; P <.001). AF and HF occurred most often, with the lifetime cumulative incidence of malignant ventricular arrhythmias occurring in 32% (95% CI, 0.23-0.40) in patients younger than 40 years at the age of diagnosis and in 1% (95% CI, 0.01-0.02) in patients older than 60 years.2
Currently, there is no cure for HCM. Although pharmacologic therapy, devices, and surgical management effectively prolong lifespan and treat symptomatology of HCM, substantial costs accompany these interventions.1,3-10
High Costs of HCM
Genetic Testing
Evaluation of familial inheritance, including a 3-generation family history, is recommended by the HCM treatment guidelines as part of the initial assessment. Family history may be pertinent to the recommendation of subsequent genetic screening of the patient and at-risk family members. The clinical benefits of genetic testing in HCM include confirmation of the diagnosis, cascade genetic testing in family members, and guidance regarding reproductive decisions. Cascade genetic testing in family members identifies the individual carrying the pathogenic variant requiring ongoing clinical surveillance while releasing those without from lifelong surveillance.1
The cost for genetic testing ranges from $3000 to $3800 for the first family member and $250 for other family members if a mutation is identified.3 Unfortunately, there is little consensus among medical payers in the United States on coverage for the genetic screening tests. In addition, different payers will include different panels for the same condition. Often, the medical policy, if it exists and is available to clinicians and patients, the requirement of “medical necessity” is often not determined until after a claim has been submitted.11
An Australian study investigating the cost-effectiveness of genetic and clinical screening compared with clinical screening alone concluded that the addition of genetic testing was a very cost-effective strategy with an incremental cost-effectiveness ratio of 587 Euro per quality-adjusted life-year (QALY) gained and 9509 Euro per additional life-year gained as determined by a probabilistic Markov decision model.12
Hospitalizations
To evaluate the financial burden of oHCM on the healthcare system, a retrospective cohort study examined National In-patient Sample (NIS) records of 2605 patients admitted to the hospital with oHCM in the United States in 2013. Thirty-three percent of the patients included in the analysis were older than 64 years, 18% were younger than 45 years, and most were female (55%). The average stay was 4.9 days, with the majority (81.19%) of patients treated in urban teaching hospitals. The study defined hospital charges as the amount the hospital charged for the entire hospital stay, excluding professional fees. Researchers converted total charges to costs using cost-to-charge ratios based on hospital accounting reports from the Centers for Medicare & Medicaid Services (CMS). The mean and median cost of admission was $25,433 and $19,422, respectively. The actual mean and median hospital charges were $88,646 and $58,460, respectively. Comparatively, this was much higher than the reported 2014 US healthcare spending of $9523 per person when the study was conducted. Private insurance paid for approximately 43% of the hospitalizations, Medicare for 37%, and Medicaid for 10%. The percentage of routine discharge was inversely proportional to age, declining from 90% to 65% from age 18 to 85. Most patients were discharged to home (76.39%), but a substantial remainder required further institutionalized care (13.05% home health care, 4.61% nursing home care, and 3.45% short-term hospital care).4
Another study specifically examined the impact of arrhythmias in patients with HCM on the cost of hospitalization and mortality. Using NIS data of 225,816 hospitalizations of patients with HCM from 2003 to 2014, the study found a 10.5% relative increase (P <.001) in the number of hospitalizations related to arrhythmias in patients with HCM, especially in patients with more comorbidities and aged 80 years or older. The total mean cost of care in patients with HCM and arrhythmia after adjusting for inflation rose from $16,105 in 2003 to $19,310 in 2014 (P-trend, <.001), representing an absolute increment in annual national cost from $125 million in 2013 to $162 million in 2014 (P-trend, <.001). The overall mean cost of HCM hospitalization with arrhythmia was $20,522 versus $15,636 with no arrhythmia, with ventricular fibrillation/flutter (VF/VFL) ($39,108) and ventricular tachycardia ($28,996). In patients with HCM and arrhythmias, in-hospital mortality was higher than those without arrhythmias. Still, there was a significant decrease in mortality rate in patients with HCM and arrhythmias from 6.2% in 2003 to 3.4% in 2014 (P-trend, <.001). In patients with HCM and arrhythmias, the mortality rate was highest in patients aged 80 years or older (7.4%), females (5.5%), and in Black (4.64%).5
Symptomatic Treatment
Alcohol Septal Ablation (ASA) or Septal Myectomy (SM)
Patients with HCM and symptomatic LVOTO refractory to medications may require invasive septal reduction therapy (SRT) with either ASA or SM. Each intervention is effective but has certain associated risks and benefits (Table1). Complete heart block as a result of SRT may require a permanent pacemaker in some patients post-procedure.1 In 2014, the mean hospital charge in the United States associated with a pacemaker was $83,521, with an in-hospital death rate of 1.46% and a mean length of hospital stay of 5.1 days.8


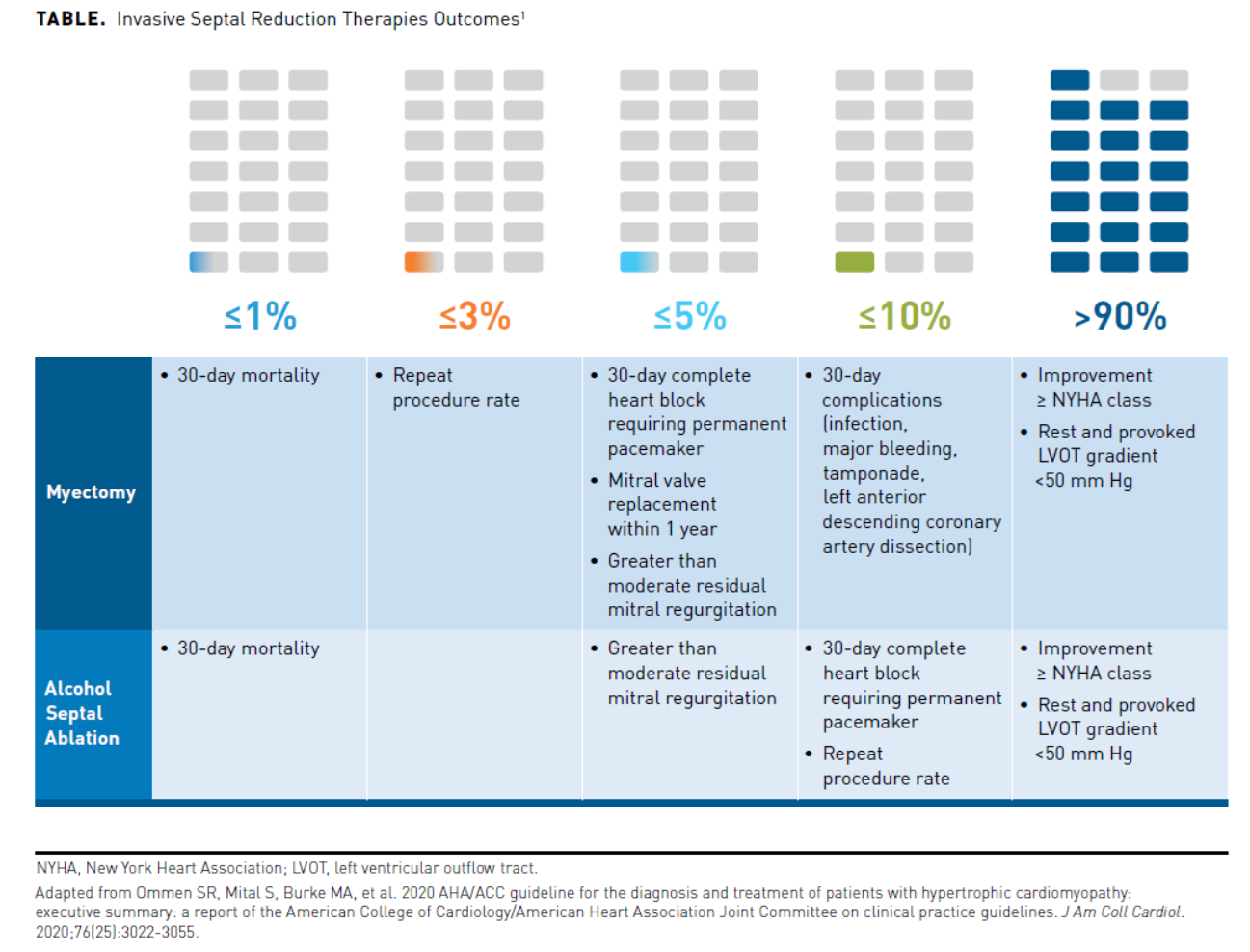
A study using the US National Readmission Database compared resource utilization, in-hospital mortality, and complication rates between the 2 age groups of patients with HCM undergoing ASA or SM. A total of 2245 patients with HCM underwent ASA (1272 patients ≤65 years, 973 patients >65 years) and 2113 patients SM (1739 patients ≤65 years, 374 patients >65 years) from 2010 to 2015. The median length of hospital stay was significantly longer (SM, 6 days vs ASA, 3 days; P <.001) in both age groups, and the total median hospital costs were greater (≤65 years: SM, $28,459 vs ASA, $13,847; P <.001; >65 years: SM, $32,429 vs ASA, $15,343; P <.001) in both age groups receiving SM versus ASA. The in-hospital mortality in patients undergoing SM was significantly greater in both age groups compared with patients undergoing ASA (≤65 years: SM, 1.5% vs ASA, 0.3%; OR, 6.2; 95% CI, 1.01-38.00; P = .048) (>65 years: SM, 6.7% vs ASA, 1.7%; OR, 4.29; 95% CI, 1.03-17.9; P = .046).6
Although the initial resource utilization and costs were lower with ASA than SM, post-procedural healthcare utilization and costs associated with ASA may increase over time compared with SM. Another study examined claims data from the OptumLabs Data Warehouse of adult patients with oHCM from 2006 to 2018, comparing the 2-year cumulative post-procedural costs and healthcare utilization after SM and ASA. A total of 379 patients underwent SM and 185 ASA, with 53% being male and of the mean age of 56.3. There was a statistically insignificant increase in the 2-year mean number of physician office visits (20 SM vs 23.4 ASA) or emergency department visits (IRR, 1.16; 95% CI, 0.76-1.77) in patients undergoing ASA versus SM. The 2-year risk of hospital readmission was similar between the 2 groups (36.9% following SM and 43.8% following ASA). There was a nonsignificant increase in readmissions in the ASA group versus the SM group (IRR 1.54; 95% CI, 0.99-2.40). The median cumulative 2-year post-procedural costs were higher in those receiving ASA ($22,165 [range, $8848-$81,143]) versus SM ($20,170 [range, $10,006-$44,755]). Rehospitalization accounted for approximately 20% (for ASA) and 17% (for SM) of these post-procedural costs.7
Implantable Cardiac Defibrillator and Short Atrioventricular Delay Pacing
An ICD has proven efficacy in aborting potentially life-threatening ventricular tachyarrhythmias and preventing SCD in patients with HCM. HCM guidelines recommend using ICD as primary prevention based on risk factors and secondary prevention in individuals surviving a cardiac arrest or hemodynamically significant ventricular fibrillation/ventricular tachycardia.1 An ICD system can deliver anti-tachycardia pacing, bradycardia pacing, cardioversion, and can be combined with cardiac resynchronization therapy when advised.9 The mean hospital charges in the United States in 2014 for an implantable defibrillator were $171,476, with an in-hospital death rate of 0.69% and a mean length of stay of 6.3 days.8
A Swedish study simulated the cost-effectiveness of an ICD over 12 years in terms of costs per life saved and cost per gained QALY versus treatment without an ICD in adults with HCM. The mean age of ICD implantation was 51.8 years based on data from a nationwide HCM cohort. The assumed willingness-to-pay for a gained QALY was 53,050 Euro. The number of lives saved was 402 of the 1000 simulated patients after 12 years, and 2.5 patients needed to be treated during the 12 years to save one life. The cost per life saved is approximately 57,117 Euro, and the incremental cost for a gained QALY was 15,150 Euro. From the societal perspective, including effects on productivity losses, in terms of QALYs, ICD treatment was superior and less expensive.9
Short atrioventricular delay-pacing is a treatment alternative to SRT for the treatment of LVOTO in oHCM. The long-term-hemodynamic results associated with short atrioventricular delay-pacing are not inferior to myectomy, but no long-term randomized studies comparing it with SRT in comparable patient groups exist. A Swedish case-control study compared the outcomes of 31 pairs of patients with oHCM treated with myectomy or pacing from 2002 to 2013. Both treatments resulted in significant improvements in LVOT-gradients as well as NYHA class. The mean cost of hospitalization was significantly less for pacing than myectomy: 74,000 (≈$11,111 in 2013 USD) ± 16,000 (≈$2,402 in 2013 USD) Swedish krona (SEK) for pacing and 310,000 (≈$46,546 in 2013 USD) ± 180,000 (≈$27,027 in 2013 USD) SEK for myectomy, P <.001), with a significantly shorter in-hospital stay (2 days vs 7 days). The myectomy group had significantly more peri-procedural complications than the pacing group (35.5% vs 3.2%, respectively; P <.0001). A pacemaker was implanted in 9.7% of patients peri-operatively and in 25.8% in late-follow up in the myectomy group. Freedom from re-interventions was observed in 61.3% in the myectomy group versus 90.3% in the pacing group (P = .003)10,13
Cardiac Transplantation (CT)/Ventricular Assist Devices (VADs)
A specific subset of patients with HCM may experience severe refractory congestive HF that ultimately requires CT. About 3.5% of patients may progress to a burnt-out end-stage distinguished by fibrotic replacement and chamber remodeling, causing systolic dysfunction and restrictive ventricular filling with a mean 3-year survival time. A heart transplantation is a life-saving intervention in patients with end-stage HCM. It is the only option to restore functional status and longevity among HCM patients with advanced HF.14 However, CT is very costly and has associated risks and morbidities. The mean hospital charges in the United States in 2014 for CT were $808,770, with an in-hospital death rate of 7.84% and mean length of stay of 45.4 days.8 Heart transplantation also requires the lifetime use of immunosuppressant medications. About 10% of heart transplant recipients need renal replacement therapy or transplant due to calcineurin inhibitor-induced nephrotoxicity. Right HF and biventricular failure may occur early postoperatively. Heart transplant recipients may also experience rejection and infectious complications. Coronary vasculopathy, requiring re-transplantation, develops in about 30% to 40% of heart transplant recipients within 5 years. Malignancy post-transplantation accounts for 10% to 23% deaths post-transplantation.14 A study of evaluating outcomes in 535 adult HCM patients in the United States registered in the Scientific Registry of Transplant recipients with HCM awaiting a heart transplant between April 2008 and October 2016 determined the waitlist mortality was 9.6%. The 12-month heart transplant rate was 64.8%, and post-transplant survival in patients with HCM was 91.6% at one year and 82.5% at 5 years.15 Bridge to transplantation (BTT) with an implantable VAD may be used in patients with HCM, but is difficult because of placement challenges due to small left ventricular chamber size, the presence of LVH (mid and distal left ventricular), and left ventricular muscle bundles, which could impede proper functioning. By report, VAD placement in patients with HCM may increase susceptibility to right ventricular dysfunction and hypotension. Many patients with HCM may not qualify for VAD placement, which may place them at a lower priority on the transplant list.14
Health-related Quality of Life (HRQOL)
In addition to the economic costs accompanying treatment interventions for HCM, reduced HRQOL in patients living with HCM has been demonstrated in studies. The Kansas City Cardiomyopathy Questionnaire (KCCQ) is a self-administered validated questionnaire measuring the perceived burden of HF symptoms. Scores range from 0 to 100, with higher values representing better HRQOL. A study in 91 patients with HF and HCM evaluated the HRQOL (57% males; median age 58) and found an overall moderate HRQOL impairment (median global KCCQ score, 67). NYHA class and the median global KCCQ score were inversely correlated (Kendall’s tau b coefficient r−0.33; P = .001). There were also significantly worse HRQOL scores in patients with pulmonary hypertension (resting pulmonary artery systolic pressure ≥45 mm Hg) than those without (median KCCQ score, 56.2 vs 77.5; P = .013). Patients with a worse HRQOL score were older and had more severe functional impairment. These included a history of syncope, poor renal function, echocardiographic parameters of increased severity and/or dysfunction, and elevated pulmonary artery systolic pressure. The study identified estimated glomerular filtration rate and NYHA functional class as independent predictive factors of the HRQOL score.16
A study conducted between 2015 and 2018 used a conceptual model to capture patients’ perspectives on the symptoms of HCM they were experiencing and their impact on their lives. A total of 444 participants who responded to the 80-question web survey reported symptoms of fatigue (74%), shortness of breath upon exertion (73%), lightheadedness (70%), exercise intolerance (57%), palpitations (54%), dizziness after exertion (54%), chest pain (39%), and syncope (24%). The impact of physical activity upon symptoms varied, with the majority reporting slight (42%) or marked limitation (31%) of activity, and 6% reported being unable to be physically active without discomfort per survey. Eighty-four percent of patients with oHCM reported 4 or more symptoms compared with 55% of patients with nHCM. The majority of patients (70%) experienced symptoms somewhat or significantly worsening since diagnosis, having a more significant impact on their ability to work. The most commonly reported effects of HCM elicited during the concept elicitation interviews of 27 patients included emotional impacts (78%), restrictions of physical activities (78%), feelings of depression and anxiety (78%), and limitations on work (63%).17
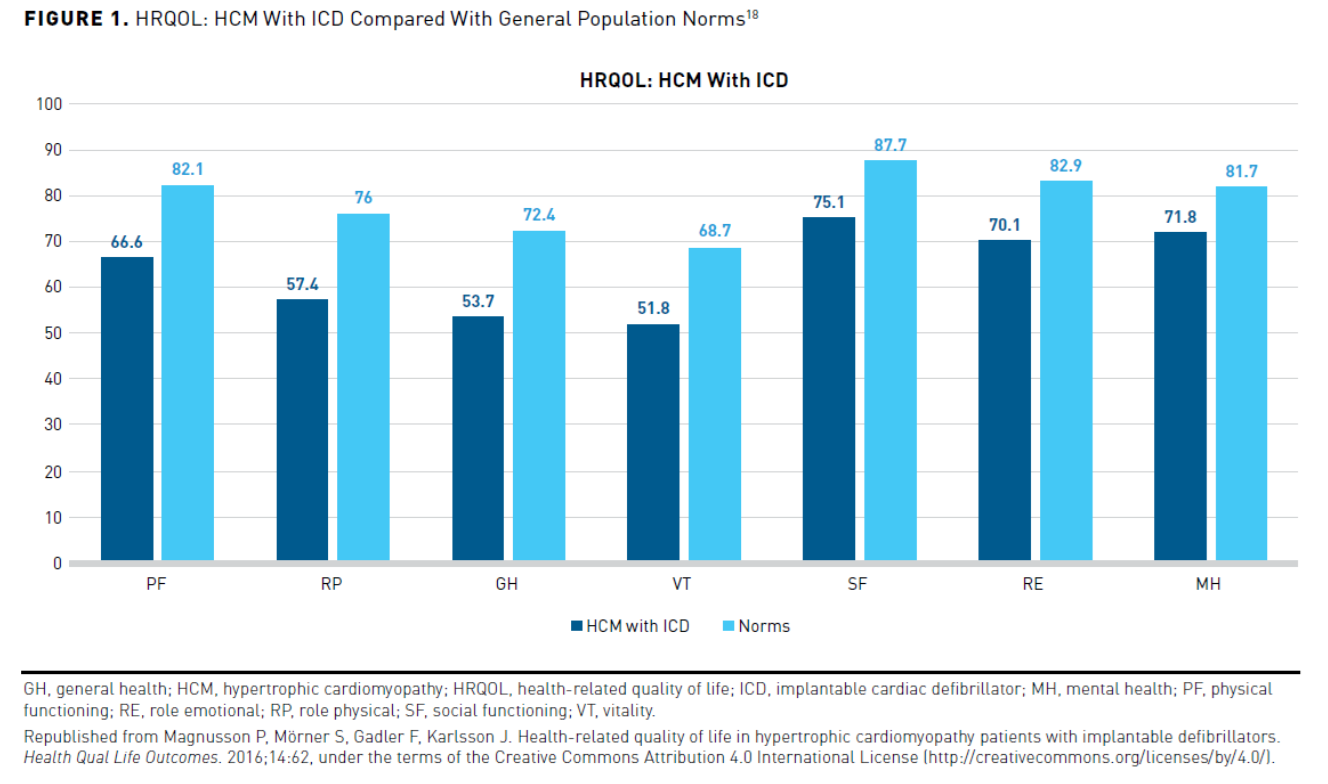
HRQOL has also been shown to be impacted in patients with HCM and an ICD in a Swedish study. The study used a validated self-reported questionnaire (36-item short form). It analyzed the responses of 245 patients with HCM and an ICD, comparing responses with a Swedish age- and sex-matched control group. The physical functioning, role physical, general health, vitality, social functioning, role emotional, mental health, physical component summary, and mental component summary scores were significantly lower (P <.001) than control (Figure 118). The most considerable impact occurred in the general health domain, with a more significant effect seen in the physical component summary scores than the mental component summary scores. Subgroup analyses showed that AF or HF significantly impacted physical functioning, physical role, and general health. AF also had a significant impact on social functioning scores. Inappropriate ICD shock negatively impacted mental health while appropriate ICD therapy trended oppositely. The HRQOL was similar between primary and secondary prevention.18


Improving Patient Outcomes
Crucial to improving patient outcomes is shared decision making. Adherence to recommendations can be enhanced when there is patient involvement in the treatment decision-making process. There should be a dialogue between patients and their healthcare providers informing them of the available testing and treatment options, including discussing all the risks and benefits. A patient should be encouraged to express their goals and concerns. The selection of interventions, including genetic evaluation, activity, lifestyle, and therapy choices, should be based on individual values, preferences, and associated conditions and morbidities. Judicious use of exercise, when advised, can also improve outcomes. Referral of patients to multidisciplinary HCM centers may help improve patient outcomes. Providers at HCM centers have specific training and competencies necessary to manage these patients’ specialized and complex needs. Particularly with invasive SRT (ASA or SM), treatment guidelines recommend that patients have the procedure performed at comprehensive or primary HCM centers with specialized training and sufficient volumes to ensure optimal outcomes.1
Emerging Therapies: Cardiac Myosin Inhibitors
Currently, no medical treatments that modify the pathology and disease course exist for HCM. Gene therapy with clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeat-associated 9 (CRISPR/Cas9), exon-skipping, trans-splicing, gene replacement, or small interfering RNAs (siRNAs) may play a future role in the treatment of HCM.19 Trials investigating small molecules, such as cardiac myosin inhibitors, in patients with HCM have shown promise. The phase 2 trial (REDWOOD-HCM) with CK274 is currently ongoing.20 Cardiac myosin inhibitors selectively inhibit cardiac myosin ATPase and reduce myocardial contractility by decreasing the available number of actin-myosin cross-bridges. The reduction in contractility reduces the outflow gradient, which may improve symptoms.19
On January 11, 2021, the FDA granted CK274 an orphan drug designation for the treatment of symptomatic HCM.21 Investigations with the cardiac myosin inhibitor mavacamten completed phase 2 clinical trial testing (MAVERICK-HCM) in patients with symptomatic nHCM and phase 3 clinical trial testing (EXPLORER-HCM) in patients with symptomatic oHCM.22,23 On July 23, 2020, the FDA granted breakthrough therapy designation to mavacamten to treat symptomatic oHCM with a New Drug Application (NDA) submission expected in the first quarter of 2021.24 An ongoing long-term extension study, MAVA-LTE, evaluating the long-term safety of mavacamten is estimated for completion in November 2025.25 These investigational therapies that target the underlying pathophysiology of HCM are not approved by the FDA yet.21,26
Managed Care Considerations
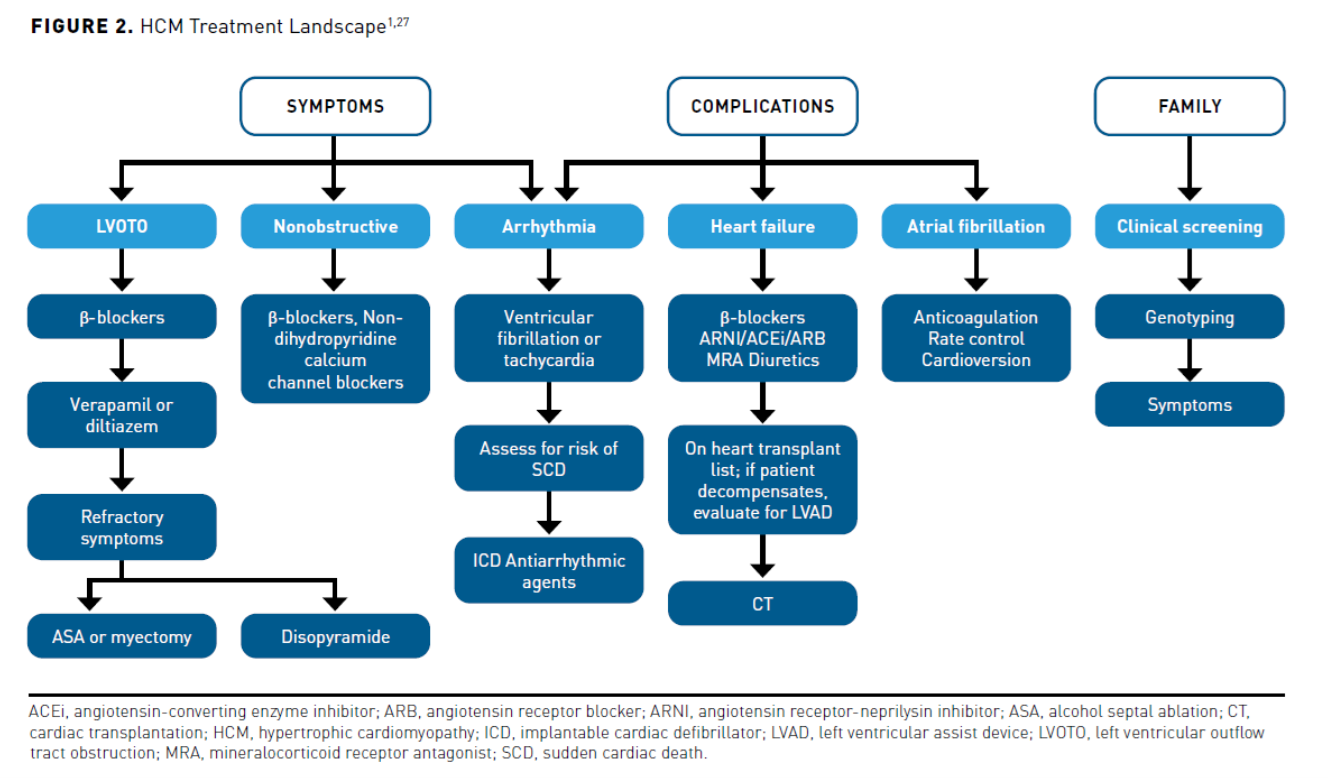
Currently, medication therapy in patients with HCM is first-line for symptomatic control (see Figure 21,27). The recommended management of HCM includes the use of low-cost generically available cardiac medications such as non-dilating β-blockers, non-dihydropyridine calcium channel blockers, and disopyramide. Other drugs may be used to treat HCM-associated complications, including anticoagulants for AF, antiarrhythmics for arrhythmias, and angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, angiotensin receptor-neprilysin inhibitors, mineralocorticoid receptor antagonists, and diuretics to treat HF. Patients refractory to treatment may require an ICD, SRT, or CT to treat symptoms and complications. See Figure 2 for current management of HCM.1,27


As new treatments are approved, further clinical evidence regarding their safety and efficacy is gathered before place in therapy is determined by clinical practice guidelines. Expedited FDA approval is based on data from clinical studies conducted over a shorter timeframe, sometimes raising concerns of long-term safety and efficacy.28 This poses challenges for managed care professionals to allocate healthcare resource utilization responsibly and effectively. The development of informed and updated formulary strategies to treat HCM is essential to integrate pharmaceutical advances based on improved risk-benefit profiles of investigational agents.29 A clinically rigorous analysis of current clinical trial data should be conducted, looking for meaningful improvement on clinically significant end points, including hospitalization and mortality, as well as hemodynamic parameters.30,31 A review of available literature, including clinical treatment guidelines, pharmacoeconomic studies, and outcomes data, is essential. Other important points of consideration include analysis of FDA prescribing information and safety data, drug and total healthcare cost data, and efficacy and cost comparisons to standard of care. Comparative effectiveness data, if available, may be a valuable resource to evaluate the use of drug versus alternative treatment options. An effective formulary management process enables the managed healthcare system to discriminate between a clinically superior and a marginally effective drug, minimizing overall medical cost and promoting patient access to affordable care.29
Utilization management strategies such as step therapy, quantity limits, and prior authorization (PA) may complement formulary management. Evidence-based PA criteria are crucial to ensure patients receive the most appropriate medication for their condition while reducing unnecessary drug use and cost. This will be more evident as new agents enter the marketplace, but even among current generically available agents, there are opportunities for enhanced utilization management to influence usage to less expensive generic agents.32
Conclusions
HCM is a disease associated with substantial economic and clinical burdens. Although improvements in earlier diagnosis, risk stratification, family screening, pharmacologic therapy, devices, and surgical management have led to better outcomes, HCM has been associated with a decreased HRQOL and psychological well-being. Thus far, management of HCM has been symptomatic and does not treat the underlying illness. Cardiac myosin inhibitors have recently shown promise in clinical trials as potential first-in-class medications to treat the underlying pathology of HCM. Managed care pharmacists should be prepared to assess their value to improve patients’ length and quality of life affected by HCM and reduce medical costs for these patients. Ultimately, the results of long-term safety and efficacy studies showing reductions in hospitalizations, morbidity, and mortality compared with standard of care will determine these agents’ value and place in practice.
Author affiliation: Patty Taddei-Allen, PharmD, MBA, BCACP, BCGP, is the Vice President, Clinical Programs and Services, WellDyne, Lakeland, FL.
Funding source: This activity is supported by an educational grant from MyoKardia, Inc, a wholly owned subsidiary of Bristol Myers Squibb.
Author disclosure: Dr Taddei-Allen has no relevant financial relationships with commercial interests to disclose.
Author information: Concept and design; drafting of the manuscript; and critical revision of the manuscript for important intellectual content.
Address correspondence to: ptaddei-allen@welldyne.com
Medical writing and editorial support provided by: Lori A. Uildriks, PharmD
References
1. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020;76(25):3022-3055. doi: 10.1016/j.jacc.2020.08.044
2. Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138(14):1387-1398. doi: 10.1161/CIRCULATIONAHA.117.033200
3. Genetic testing for hypertrophic cardiomyopathy: special considerations. Hypertrophic Cardiomyopathy Association. Accessed January 24, 2021. www.4hcm.org/content.asp?contentid=187#:~:text=If%20a%20person%20chooses%20to,rate%20that%20is%20around%20%24250
4. Jan A, Shah MA, Rehman S, Rungatscher A, Ahmed N, Faggian G. Hypertrophic obstructive cardiomyopathy and the cost of treatment. EJCM. 2016;4(2):27-32. doi: 10.15511/ejcm.16.00227
5. Tripathi B, Khan S, Arora S, et al. Burden and trends of arrhythmias in hypertrophic
cardiomyopathy and its impact of mortality and resource utilization. J Arrhythm. 2019;35(4):612-625. doi: 10.1002/joa3.12215
6. Lemor A, Villablanca PA, Hosseini Dehkordi SH, et al. Comparison of outcomes of alcohol septal ablation or septal myectomy for hypertrophic cardiomyopathy in patients ≤65 years versus >65 years. Am J Cardiol. 2020;127:128-134. doi: 10.1016/j.amjcard.2020.04.018
7. Sun D, Schaff H, Van Houten HK, et al. Health care utilization and costs of surgical septal myectomy and alcohol septal ablation for obstructive hypertrophic cardiomyopathy. CF2 Abstract. Western Thoracic Surgical Association 46th Annual Meeting. June 24-27, 2020. Accessed March 31, 2021. https://meetings.westernthoracic.org/abstracts/2020/CF2.cgi
8. Virani SS, Alonso A, Benjamin EJ, et al; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics−2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139-e596. doi: 10.1161/CIR.0000000000000757
9. Magnusson P, Wimo A. Health economic evaluation of implantable cardioverter defibrillators in hypertrophic cardiomyopathy in adults. Int J Cardiol. 2020;311:46-51. doi: 10.1016/j.ijcard.2020.02.055
10. Javidgonbadi D, Andersson B, Abdon NJ, Östman-Smith I. Morbidity and resource usage after myectomy- or pacing-treatment in hypertrophic obstructive cardiomyopathy: a case-control study. Int J Cardiol. 2021;322:197-203. doi: 10.1016/j.ijcard.2020.08.066
11. Spoonamore KG, Johnson NM. Who pays? Coverage challenges for cardiovascular genetic testing in US patients. Front Cardiovasc Med. 2016;3:14. doi: 10.3389/fcvm.2016.00014.
12. Ingles J, McGaughran J, Scuffham PA, Atherton J, Semsarian C. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart. 2012;98(8):625-630. doi: 10.1136/heartjnl-2011-300368
13. PoundSterlingLive.com. Historical rates for the USD/SEK currency conversion on November 13, 2013. Accessed March 27, 2021. poundsterlinglive.com/best-exchange-rates/us-dollar-to-swedish-krona-exchange-rate-on-2013-11-13
14. Torres MF, Perez-Villa F. Heart transplantation in patients with hypertrophic cardiomyopathy. Glob Cardiol Sci Pract. 2018;2018(3):32. doi: 10.21542/gcsp.2018.32
15. Zuñiga Cisneros J, Stehlik J, Selzman CH, Drakos SG, McKellar SH, Wever-Pinzon O. Outcomes in patients with hypertrophic cardiomyopathy awaiting heart transplantation. Circ Heart Fail. 2018;11(3):e004378. doi: 10.1161/CIRCHEARTFAILURE.117.004378
16. Capota R, Militaru S, Ionescu AA, et al. Quality of life status determinants in hypertrophic cardiomyopathy as evaluated by the Kansas City Cardiomyopathy Questionnaire. Health Qual Life Outcomes. 2020;18(1):351. doi: 10.1186/s12955-020-01604-9
17. Zaiser E, Sehnert AJ, Duenas A, Saberi S, Brookes E, Reaney M. Patient experiences with hypertrophic cardiomyopathy: a conceptual model of symptoms and impacts on quality of life. J Patient Rep Outcomes. 2020;4(1):102. doi: 10.1186/s41687-020-00269-8
18. Magnusson P, Mörner S, Gadler F, Karlsson J. Health-related quality of life in hypertrophic cardiomyopathy patients with implantable defibrillators. Health Qual Life Outcomes. 2016;14:62. doi: 10.1186/s12955-016-0467-x
19. Repetti GG, Toepfer CN, Seidman JG, Seidman CE. Novel therapies for prevention and early treatment of cardiomyopathies. Circ Res. 2019;124(11):1536-1550. doi: 10.1161/CIRCRESAHA.119.313569
20. Randomized evaluation of dosing with CK-3773274 in obstructive outflow disease in HCM (REDWOOD-HCM). ClinicalTrials.gov. Updated December 11, 2020. Accessed January 24, 2021. clinicaltrials.gov/ct2/show/NCT04219826?cond=REDWOOD-HCM&draw=2&rank=1
21. Cytokinetics granted orphan drug designation for CK-3773274 for the treatment of hypertrophic cardiomyopathy. News release. Published January 11, 2021. Accessed January 24, 2021. www.globenewswire.com/news-release/2021/01/11/2156166/0/en/Cytokinetics-Granted-Orphan-Drug-Designation-for-CK-3773274-for-the-Treatment-of-Hypertrophic-Cardiomyopathy.html
22. A phase 2 study of mavacamten in adults with symptomatic non-obstructive hypertrophic cardiomyopathy (nHCM) (MAVERICK-HCM). ClinicalTrials.gov. Updated October 19, 2020. Accessed January 24, 2021. clinicaltrials.gov/ct2/show/NCT03442764?term=MAVERICK-HCM&draw=2&rank=2
23. Clinical study to evaluate mavacamten (MYK-461) in adults with symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM). ClinicalTrials.gov. Updated November 17, 2020. Accessed January 24, 2021. clinicaltrials.gov/ct2/show/NCT03470545
24. MyoKardia announces receipt of breakthrough therapy designation from FDA for mavacamten for the treatment of symptomatic, obstructive hypertrophic cardiomyopathy. Press release. Bristol Myers Squibb. Published July 23, 2020. Accessed January 24, 2021. myokardia.gcs-web.com/news-releases/news-release-details/myokardia-announces-receipt-breakthrough-therapy-designation-fda
25. A long-term safety extension study of mavacamten in adults who have completed MAVERICK-HCM or EXPLORER-HCM. ClinicalTrials.gov. Updated November 18, 2020. Accessed January 24, 2021. clinicaltrials.gov/ct2/show/NCT03723655?term=explorer+hcm&draw=2&rank=1
26. Olivotto I, Oreziak A, Barriales-Villa R, et al; EXPLORER-HCM study investigators. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial [Erratum in: Lancet. 2020;396(10253):758]. Lancet. 2020;396(10253):759-769. doi: 10.1016/S0140-6736(20)31792-X
27. Jacoby DL, DePasquale EC, McKenna WJ. Hypertrophic cardiomyopathy: diagnosis, risk stratification and treatment. CMAJ. 2013;185(2):127-134. doi: 10.1503/cmaj.120138
28. Richardson E. Breakthrough therapy designation: The FDA has new authority to expedite the approval process for drugs intended to treat serious or life-threatening conditions. Health Affairs. May 15, 2014. Accessed January 24, 2021. healthaffairs.org/do/10.1377/hpb20140515.807039/full/healthpolicybrief_115.pdf
29. Formulary management. Academy of Managed Care Pharmacy. July 18, 2019. Accessed January 24, 2021. amcp.org/about/managed-care-pharmacy-101/concepts-managed-care-pharmacy/formulary-management
30. Healy M, Goldman M. Critical analysis of mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomized, double-blind, placebo-controlled, phase 3 trial. Hurst’s the Heart Updates. AccessCardiology. McGraw-Hill; 2020. Accessed January 24, 2021. accesscardiology.mhmedical.com/updatesContent.aspx?gbosid=553250§ionid=250281145
31. Elliott PM. The end of the beginning for drug therapy in obstructive hypertrophic cardiomyopathy with EXPLORER-HCM. Cardiovasc Res. 2020;116(13): e175-e178. doi: 10.1093/cvr/cvaa282
32. Prior authorization. Academy of Managed Care Pharmacy. July 18, 2019. Accessed January 24, 2021. amcp.org/about/managed-care-pharmacy-101/concepts-managed-care-pharmacy/prior-authorization

