
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
2022 ESC/ERS Guideline-Recommended Management of Pulmonary Arterial Hypertension and Agents Under Clinical Development
In patients with pulmonary hypertension (PH), high blood pressure occurs in the vessels between the lungs and heart, resulting in increased muscle development in the vessel walls and greater resistance in the pulmonary arteries.1 Heart failure (HF) is among the most severe downstream effects of PH.2 Pulmonary arterial hypertension (PAH) is a subtype of PH defined as a mean pulmonary artery pressure of at least 25 mm Hg, mean pulmonary artery pressure (PAP) of at least 25 mm Hg, and pulmonary artery occlusion pressure of no more than 15 mm Hg.3,4 Idiopathic PAH is believed to be the most common form of PAH (50%-60% of all cases).5
Although PAH remains difficult to diagnose and treat, the clinical application of results from recent clinical studies has changed the way that the disease is diagnosed and managed, as reflected in the revised guidelines for managing PAH jointly developed in 2022 by the European Society of Cardiology (ESC)/European Respiratory Society (ERS). These guidelines were endorsed by the International Society for Heart and Lung Transplantation and the European Reference Network on rare respiratory diseases.5 Proceedings from the Sixth World Symposium on Pulmonary Hypertension that were retained in the 2022 ESC/ERS guidelines were used to develop a treatment algorithm based on risk stratification in patients with PAH.6 The following treatment recommendations were outlined in the proceedings6:
- In treatment-naïve patients with PAH, initial monotherapy improves exercise capacity, hemodynamics, and outcomes when compared with those noted among untreated patients.
- In patients who are newly diagnosed with PAH and treatment-naïve, initial combination therapy can improve symptoms, exercise capacity, and outcomes compared with those in individuals who receive initial monotherapy.
- In patients with PAH already receiving treatment, sequential combination therapy can improve exercise capacity, hemodynamics, and outcomes compared with the effects of background therapy alone.
Numerous available and emerging approaches can be used to align with the guideline recommendations.
Overview of Available PAH Treatments
Pharmacologic Therapies
Currently available agents for treating PAH target the NO-sGC-cGMP, endothelin, or prostacyclin (PGI2) pathways (Figure).5 Although the use of these therapies has been associated with improved survival in patients with PAH in recent years, their specific impact and those of related treatment strategies require further study.5


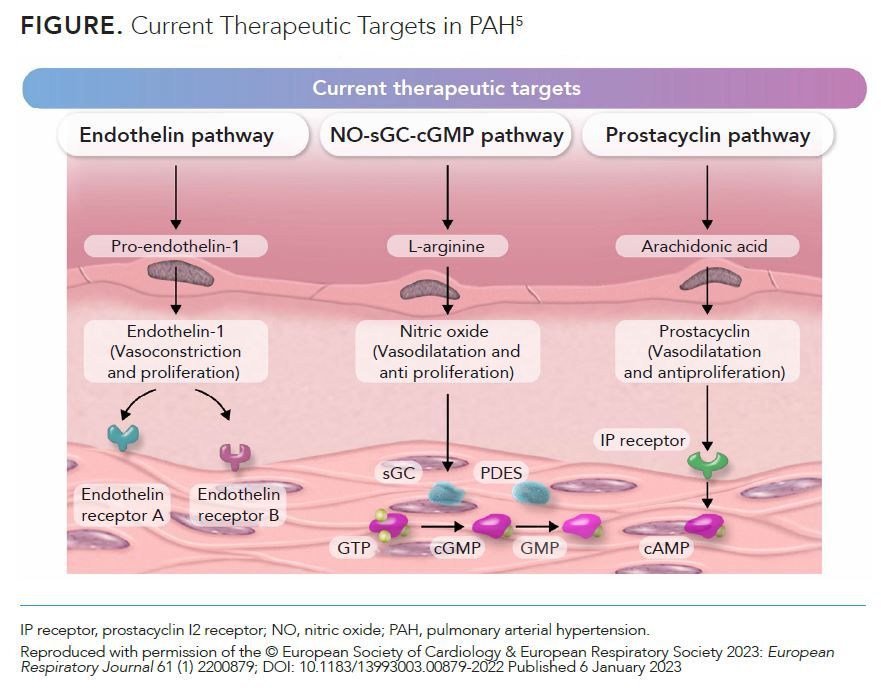
Calcium Channel Blockers
Vasoreactivity testing can be performed to determine how patients with idiopathic, heritable, or drug- or toxin-induced PAH respond to vasodilator challenge. This method is not indicated for patients with other forms of PAH, as they typically do not exhibit vasoreactivity.7 The results of this test can help guide treatment selection, as patients with PAH who show a positive response in acute vasoreactivity testing may benefit from calcium channel blocker (CCB) therapy with nifedipine, diltiazem, felodipine, or amlodipine.5,8 CCBs inhibit vasoconstriction by preventing the influx of calcium ions into vascular cells.9 However, Sitbon and colleagues showed that less than 10% of patients with idiopathic PAH respond to CCBs, highlighting the need for advanced therapies.10
Endothelin Receptor Antagonists
The exercise capacity of patients with PAH may be increased by treatment with endothelin receptor antagonists (ERAs) (ambrisentan, bosentan, and macitentan).5 Drugs in the PDE5 inhibitor (PDE5i) and soluble guanylate cyclase (sGC) stimulator classes may improve exercise capacity, symptoms, and hemodynamics in patients with PAH.5 Therapies supported by the ESC/ERS guidelines include the PDE5is sildenafil and tadalafil and sGC stimulator riociguat. Treatment with certain PDE5is also lowers the PAP and pulmonary vascular resistance (PVR) and improves the cardiac index of patients with PAH.11
Prostacyclin Analogs and Receptor Agonists
PGI2 analogs and PGI2 receptor agonists are associated with improvement in vasodilation, inhibition of platelet aggregation, and exertion of cytoprotective and antiproliferative effects.5 Examples of drugs in these classes that are recommended for treating PAH include epoprostenol, iloprost, treprostinil, beraprost, and selexipag. Table 1 summarizes findings on available pharmaceutical agents.12-24


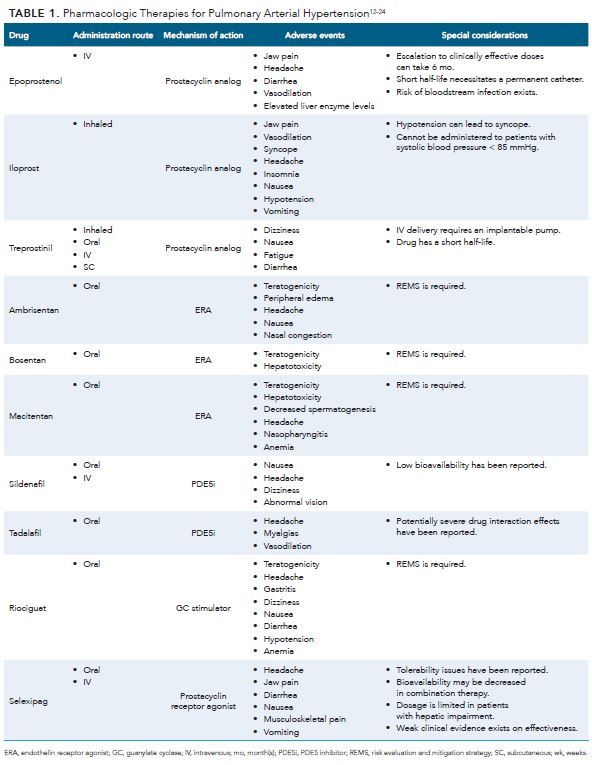
Supportive Pharmacologic Therapies
Various supportive therapies are available for treating patients with PAH. These treatments can improve disease symptoms and the quality of life (QOL) of patients.
Anticoagulation Therapy
Use of anticoagulation therapy in patients with PAH is not supported by the results of randomized controlled trials. However, the ERS/ESC guidelines highlight several reasons to consider this approach on an individual basis.5 Abnormalities in the coagulation and fibrinolytic system have been observed in patients with PAH, and in situ thrombosis of pulmonary vessels has been observed in histopathological samples from the lungs of affected patients. Patients with PAH who are prescribed anticoagulants must be followed closely because of the increased bleeding risk associated with these agents.
Diuretic Therapy
It is important for clinicians to implement measures that avoid fluid retention in patients with PAH.5 If signs of right-sided HF and edema develop, it is recommended that clinicians prescribe diuretics and restrict the patient’s fluid intake. Patients with PAH who are given diuretic therapy should be monitored closely; it is important that they regularly weigh themselves and contact their health care provider if they gain weight.
Oxygen Therapy
Although evidence for its use is sparse, oxygen therapy may benefit some patients with PAH, including those with concomitant chronic obstructive pulmonary disease or those who experience oxygen desaturation during exercise or sleep.5
Antihypertensive Drugs
The usefulness and safety of drugs for hypertension or left-sided HF in patients with PAH are unclear, and no recommendations are provided in the ESC/ERS guidelines. Importantly, these drugs can cause dangerous drops in blood pressure and/or heart rate.5
Therapy for Mineral Deficiency
Iron deficiency is common in patients with PAH; it is associated with impaired myocardial function, aggravated symptoms, and an increased risk of death. Therefore, the iron status of patients with PAH should be closely monitored.5
Supportive Nonpharmacologic Therapies
Physical activity or exercise training and supervised rehabilitation can improve the exercise capacity (as measured using the 6-minute walk distance [6MWD] test) and QOL of patients with PAH, as long as the specific functional limitations of each patient are observed.5 The ESC/ERS guidelines report that most studies on QOL included patients who were stable on medical treatment. Therefore, patients with PAH should reach a stable clinical condition before exercise training and supervised rehabilitation are prescribed. Patient-reported outcome measures are considerably underused in clinical trials of treatments for PAH.5 Validated outcome measurement tools (eg, Cambridge Pulmonary Hypertension Outcome Review, emPHasis-10 score, Living with Pulmonary Hypertension Questionnaire, and Pulmonary Arterial Hypertension-Symptoms and Impact [PAH-SYMPACT] score) should be used to assess changes in the health-related QOL of individuals with PAH as well as patients’ functional status, clinical deterioration, and prognosis.5
Patients diagnosed with PAH often develop psychological, emotional, and social issues; health care providers must address these concerns with empathy and hope. Frequent delays in diagnosis along with the development of physical limitations can lead to depression, anxiety, and adjustment disorders, particularly because PAH severely affects the ability of patients to work. Patients should be assessed using psychosocial screening tools and referred to appropriate care, including psychosocial specialists and patient support groups.5 Validated tools such as the updated REVEAL Registry Risk Score 2.0 can help health care providers and patients to make more informed decisions, and they should be applied more frequently in clinical practice.25 Simplified tools, such as the REVEAL Lite 2 Risk Calculator, can differentiate between a low, intermediate, and high risk of death in 1 year based on 6 modifiable factors (functional class, systolic blood pressure, heart rate, 6MWD, BNP/N-terminal pro-BNP level, and renal insufficiency).25 This tool is simple to implement, and may be useful in routine clinical practice.
Interventional Therapies
Several interventional therapies are available to patients with PAH when drug treatment is not successful. Balloon atrial septostomy or a Potts shunt procedure can be employed to decompress the right heart and increase systemic blood flow, thereby improving systemic oxygen transport despite arterial oxygen desaturation.5 These complex and high-risk procedures should only be performed at specialized centers. Patients with PAH and advanced right ventricular (RV) failure may require intensive treatment to manage triggers of right-sided HF (eg, infection, arrhythmias, anemia, other comorbidities).5 Fluid management is critical in these patients, as a negative fluid balance is necessary to reduce the RV preload and improve RV function. Patients may require mechanical circulatory support, which is available at specialized centers.5 Venoarterial extracorporeal membrane oxygenation, the most common therapy, is often used as a bridge to lung transplantation, but it can also be used to treat potentially reversible RV failure.
Lung transplantation is a critical option for patients with PAH who do not respond to optimal therapy.5 Early referral to a lung transplant center is necessary for patients with PAH who show a poor response to pharmacologic treatment, particularly to combination therapy, or for those who present with an intermediate to high risk of death. Most transplant patients with PAH undergo double-lung transplantation; those with untreatable cardiac conditions may undergo heart-lung transplantation.26
PAH Treatment Options in Development
Several treatments for PAH are in late-stage development (Table 2).5,27-36 Two agents, sotatercept and ralinepag, are being investigated in phase 3 trials.5,27


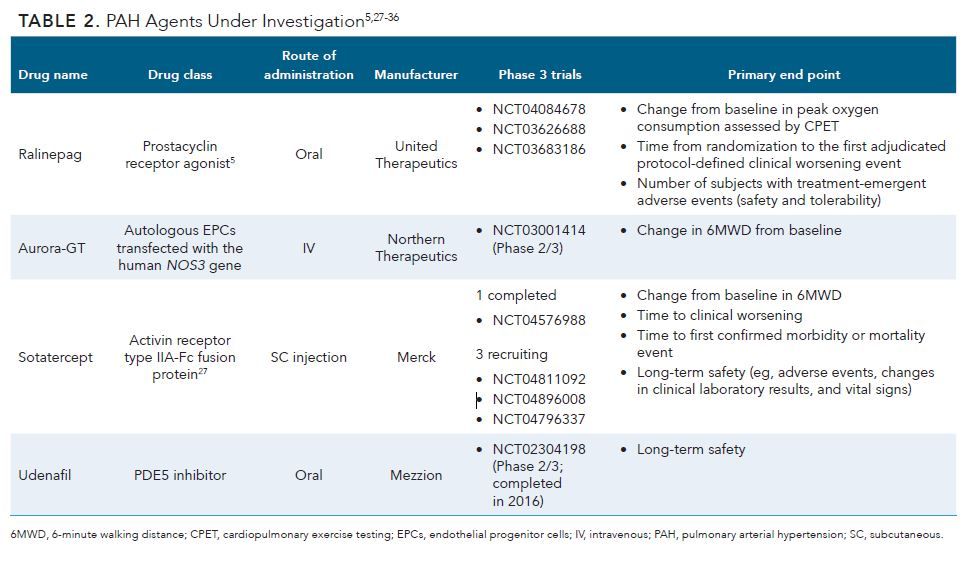
Sotatercept
Sotatercept is an activin receptor type IIA-Fc fusion protein being tested in clinical trials as a first-in-class treatment for PAH.27 It has been granted a Breakthrough Therapy Designation by the FDA. Sotatercept restores signaling mediated by ActRIIA/Smad2/3 and BMPRII/Smad1/5/8; this signaling is involved in remodeling of the pulmonary arterial wall and right ventricle.27,37
In the phase 2, multicenter PULSAR trial, 106 adults with PAH who were receiving background therapy were randomly assigned to receive sotatercept 0.3 mg/kg or 0.7 mg/kg every 3 weeks or a placebo for 24 weeks. Both doses of sotatercept were associated with significant changes in PVR (ie, the primary end point) from baseline to week 24 (decreases of 162.2 and 255.9 dyn/s/cm-5 in the 0.3-mg/kg and 0.7-mg/kg groups, respectively) (P < .01).38 Thrombocytopenia, the most common adverse event (AE) of interest, occurred in 2 patients in the 0.3-mg/kg group (6.0%), 5 patients in the 0.7-mg/kg group (12.0%), and none of the patients in the placebo group (0%).38 An increased hemoglobin level was observed as a hematologic AE in patients given sotatercept at week 24 (0.3 mg/kg: 1 patient [3%; mean (SD), 1.2 (1.2) g/dL]; 0.7 mg, 7 patients [17%; 1.5 (1.1) g/dL]; placebo: 0 patients [0%]). An increased hemoglobin level was also reported in previous trials.38,39
A total of 97 patients continued with an open-label extension of PULSAR for assessment of the long-term safety and tolerability of sotatercept.40 The safety profile of sotatercept was consistent with that recorded in prior clinical trials. Leukopenia, neutropenia, and thrombocytopenia were observed in 18 (17.3%) of 104 patients evaluated for treatment-emergent AEs (TEAEs).40 No serious TEAEs considered to be related to sotatercept use led to treatment or study discontinuation, and no deaths were attributed to use of the drug.40 Telangiectasia was observed as a new event in 11 patients (10.6%); all cases were mild, and additional monitoring is necessary to determine the clinical significance of this AE.40 Data from the placebo-controlled study and open-label extension support using a dose of 0.7 mg/kg in further phase 3 studies.40
The efficacy and safety of sotatercept for treating PAH as an add-on to background therapy (monotherapy, double therapy, or triple therapy) was evaluated in the STELLAR trial, a phase 3, randomized, double-blind, placebo-controlled, multicenter study.30 A total of 323 adult patients (mean [SD] age, 47.9 [14.8] years) with PAH were randomly assigned 1:1 to receive sotatercept or placebo via subcutaneous administration once every 3 weeks. Patients in the sotatercept group experienced a significant and clinically meaningful improvement in the primary end point of exercise capacity (as measured in by the 6MWD at 24 weeks). Changes from baseline in the 6MWD were a median of 34.4 m (95% CI, 33.0-35.5 m; P < .001) in the sotatercept group and 1.0 m (95% CI, –0.3 to 3.5 m) in the placebo group.30 Improvements were observed in 8 of 9 secondary efficacy end points, including the proportion of participants achieving multicomponent improvement (ie, improvements in 6MWD, N-terminal pro-BNP level, and either improvement in World Health Organization [WHO] functional class or maintenance of WHO functional class II) and time to death or the first clinical worsening event. Only the cognitive/emotional impacts domain score of PAH-SYMPACT did not differ significantly between the sotatercept and placebo groups. At least 10% of patients in both groups reported AEs over the 24-week treatment period, with nosebleed, telangiectasia, and dizziness being the most commonly reported. Twenty-three patients (14.1%) in the sotatercept group and 36 patients (22.5%) in the placebo group experienced serious AEs; these events led to treatment discontinuation in 3 (1.8%) and 10 (6.2%) patients, respectively.30
Ralinepag
The efficacy and safety of ralinepag, an oral PGI2 receptor agonist, are currently being tested in phase 3 trials.28 In a phase 2, double-blind, multicenter trial, 61 adult patients with PAH who were receiving monotherapy or dual background therapy were randomly assigned 2:1 to receive ralinepag or placebo for 22 weeks.41 The primary efficacy end point was the absolute change in PVR from baseline to week 22. Secondary end points included the percentage change from baseline in PVR, 6MWD, time to clinical worsening, safety, and tolerability. Ralinepag was associated with a significant reduction in the median PVR at 22 weeks compared with the effects of the placebo (P = .02). There was a 29.8% reduction in the least-squares mean PVR versus the effects of the placebo (P = .03). At week 22, the least-squares mean change from baseline in the 6MWD with ralinepag did not significantly differ from that of the placebo group (increase of 36.2 m with ralinepag vs 29.4 m with placebo). Although the percentage of patients who showed clinical worsening was lower with ralinepag (2.5%) than with placebo (9.5%), the difference was not significant (P = .26).
Almost all patients in both study groups experienced at least 1 AE. Among AEs attributed to ralinepag, 20% were mild, 47.5% were moderate, and 37.5% were severe. The most common AEs reported in the ralinepag group were headache (78%), nausea (50%), and diarrhea (48%). Ralinepag was well-tolerated in most patients during the 22-week study, and the noted AEs were consistent with those observed for other PGI2 receptor agonists.41 The rate of serious AEs was lower in the ralinepag group (10%) than that in the placebo group (28.6%). One patient in the ralinepag group experienced electrocardiographic QT prolongation as a serious AE. However, this patient had a prior condition (intraventricular conduction disturbance) that may have contributed to QT prolongation. Five patients (12.5%) withdrew from the study because of AEs that may have been related to use of the study drug; 2 patients (10%) in the placebo group withdrew from the study. The frequency of AEs decreased during the dose titration period as the dose of ralinepag was increased.
Other Therapies
A recent network meta-analysis of data from 53 randomized controlled trials of FDA-approved drugs and investigational agents from database inception through 2021 was conducted to compare the efficacies of PAH medications.42 Combination therapy with an ERA and PDE5i was associated with a reduced risk of clinical worsening (120.7 fewer events/1000 patients; 95% CI, 93.4-136.8 fewer events/1000 patients) compared with the effects of each therapy alone (51.5-85.3 fewer events/1000 patients). Riociguat therapy led to similar outcomes, but the results showed greater uncertainty (133.6 fewer events/1000 patients; 95% CI 85.3-151.3 fewer events/1000 patients).42
The long-term safety of the oral PDE5i udenafil was assessed in 18 patients with PAH in phase 2 studies.31 Both high and low doses (50 mg and 100 mg) of udenafil improved hemodynamic parameters and reduced the systolic PAP in patients with PAH while causing no serious AEs.43 Hemodynamic monitoring revealed maximum mPAP reductions of 11.0 mm Hg and 8.0 mm Hg in patients treated with 50 mg and 100 mg of udenafil, respectively, whereas the placebo group showed no significant change. In a related double-blind, placebo-controlled study of 63 patients, udenafil improved the exercise capacity of patients with PAH, as demonstrated by results from the 6MWD test (changes of 46 m vs 21 m in the udenafil vs placebo group, respectively; P = .0873).44
Gene-based therapies also are being investigated for PAH. Aurora-GT is an angiogenic cell therapy that will be investigated for PAH in a phase 2/3 trial (SAPPHIRE) at several Canadian locations.29,45 The therapy involves monthly infusions of autologous endothelial progenitor cells transfected with the human NOS3 gene to restore eNOS enzyme, which dilates the pulmonary blood vessels.
Conclusions
PAH is a rare but serious progressive condition associated with a high mortality rate. There are currently no curative treatments available, and medications only address disease symptoms. There are several unmet needs in managing PAH, including the need for earlier diagnosis, more frequent evaluation once patients receive a diagnosis, improved adherence to treatment, and treatments that control disease progression. The results of studies demonstrated improved clinical outcomes with delays in disease progression; however, currently available PAH-specific therapies do not target the specific mechanisms of the disease. New agents are currently being developed in existing drug classes; these include the PGI2 receptor agonist ralinepag being investigated in phase 3 clinical trials and the PDE5i udenafil being studied in phase 2/3 trials. The fusion protein sotatercept is being tested in phase 3 trials, and the gene therapy Aurora-GT is being evaluated in phase 2/3 trials. Recent advancements in the understanding of the molecular mechanisms of PAH have led to the development of promising therapies that may improve symptoms, delay clinical worsening, and promote a better QOL for affected patients.
References
- Pulmonary hypertension. Centers for Disease Control and Prevention. Updated December 3, 2019. Accessed February 24, 2023. https://www.cdc.gov/
heartdisease/pulmonary_hypertension.htm - Learn about pulmonary arterial hypertension. American Lung Association. Updated January 10, 2023. Accessed January 17, 2023. https://www.lung.org/lung-health-diseases/lung-disease-lookup/pulmonary-arterial-hypertension/learn-about-pulmonary-arterial-hypertension
- Dodson MW, Brown LM, Elliott CG. Pulmonary arterial hypertension. Heart Fail Clin. 2018;14(3):255-269. doi:10.1016/j.hfc.2018.02.003
- Prins KW, Thenappan T. World Health Organization Group I pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34(3):363-374. doi:10.1016/j.ccl.2016.04.001
- Humbert M, Kovacs G, Hoeper MM, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618-3731. doi:10.1093/eurheartj/ehac237
- Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. doi:10.1183/13993003.01889-2018
- Montani D, Savale L, Natali D, et al. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J. 2010;31(15):1898-1907. doi:10.1093/eurheartj/ehq170
- Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76-81. doi:10.1056/NEJM199207093270203
- Elliott WJ, Ram VS. Calcium channel blockers. J Clin Hypertens (Greenwich). 2011;13(9):687-689. doi:10.1111/j.1751-7176.2011.00513.x
- Sitbon O, Humbert M, Jaïs X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105-3111. doi:10.1161/CIRCULATIONAHA.104.488486
- Galiè N, Brundage BH, Ghofrani HA, et al; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894-2903. doi:10.1161/CIRCULATIONAHA.108.839274
- Prokes M, Root A. A retrospective analysis of adherence to risk evaluation and mitigation strategies requirements for pulmonary arterial hypertension drugs. Hosp Pharm. 2019;54(5):309-313. doi:10.1177/0018578718791509
- Lang IM, Gaine SP. Recent advances in targeting the prostacyclin pathway in pulmonary arterial hypertension. Eur Resp Rev. 2015;24(138):630-641. doi:10.1183/16000617.0067-2015
- Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26);2522-2533. doi:10.1056/NEJMoa1503184
- Olschewski H, Simonneau G, Galiè N, et al; Aerosolized Iloprost Randomized Study Group. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322-329. doi:10.1056/NEJMoa020204
- Galiè N, Olschewski H, Oudiz RJ, et al; Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010-3019. doi:10.1161/CIRCULATIONAHA.107.742510
- Waxman A, Restrepo-Jaramillo R, Thenappan T, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325-334. doi:10.1056/NEJMoa2008470
- Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896-903. doi:10.1056/NEJMoa012212
- Khadka A, Singh Brashier DB, Tejus A, Sharma AK. Macitentan: an important addition to the treatment of pulmonary arterial hypertension. J Pharmacol Pharmacother. 2015;6(1):53-57. doi:10.4103/0976-500X.149151
- Bhogal S, Khraisha O, Al Madani M, Treece J, Baumrucker SJ, Paul TK. Sildenafil for pulmonary arterial hypertension. Am J Ther. 2019;26(4):e520-e526. doi:10.1097/MJT.0000000000000766
- Henrie AM, Nawarskas JJ, Anderson JR. Clinical utility of tadalafil in the treatment of pulmonary arterial hypertension: an evidence-based review. Core Evid. 2015;10:99-109. doi:10.2147/CE.S58457
- Kenny M, Clarke MM, Pogue KT. Overview of riociguat and its role in the treatment of pulmonary hypertension. J Pharm Pract. 2022;35(3):437-444. doi:10.1177/0897190020961291
- Barst RJ, McGoon M, McLaughlin, et al; Beraprost Study Group. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119-2125. doi:10.1016/s0735-1097(03)00463-7
- Coghlan JG, Picken C, Clapp LH. Selexipag in the management of pulmonary arterial hypertension: an update. Drug Healthc Patient Saf. 2019;11:55-64. doi:10.2147/DHPS.S181313
- Benza RL, Kanwar MK, Raina A, et al. Development and validation of an abridged version of the REVEAL 2.0 risk score calculator, REVEAL Lite 2.0, for use in patients with pulmonary arterial hypertension. Chest. 2021;159(1):337-346. doi:10.1016/j.chest.2020.08.2069
- Humbert M, Lau EMT, Montani D, Jaïs X, Sitborn O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130(24):2189-2208. doi:10.CIRCULATIONHA.114.006974
- Merck announces positive top-line results from pivotal phase III STELLAR trial evaluating sotatercept for the treatment of adults with pulmonary arterial hypertension. Press release. Merck. October 10, 2022. Date Accessed January 17, 2023. https://www.merck.com/news/merck-announces-positive-top-line-results-from-pivotal-phase-3-stellar-trial-evaluating-sotatercept-for-the-treatment-of-adults-with-pulmonary-arterial-hypertension-pah
- A study evaluating the efficacy and safety of ralinepag to improve treatment outcomes in PAH patients. ClinicalTrials.gov. Updated February 1, 2023. Accessed February 24, 2023. https://clinicaltrials.gov/ct2/show/NCT03626688
- Study of angiogenic cell therapy for progressive pulmonary hypertension: intervention with repeat dosing of eNOS-enhanced EPCs (SAPPHIRE). ClinicalTrials.gov. Updated January 11, 2023. Accessed February 24, 2023.
https://clinicaltrials.gov/ct2/show/NCT03001414 - Hoeper MM, Badesch DB, Ghofrani HA, et al; STELLAR Trial Investigators. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. Published online March 6, 2023. doi:10.1056/NEJMoa2213558
- Clinical trial to evaluate the long-term safety of udenafil in patient with PAH. ClinicalTrials.gov.Updated April 18, 2016. Accessed February 24, 2023. https://clinicaltrials.gov/ct2/show/NCT02304198
- A study of ralinepag to evaluate effects on exercise capacity by CPET in subjects with WHO Group 1 PH (CAPACITY). ClinicalTrials.gov. Updated March 6, 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT04084678
- A study evaluating the long-term efficacy and safety of ralinepag in subjects with PAH via an open-label extension. ClinicalTrials.gov. Updated January 20, 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT03683186
- Study of sotatercept in newly diagnosed intermediate- and high-risk PAH participants (MK-7962-005/A011-13) (HYPERION). ClinicalTrials.gov. Updated March 7, 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT04811092
- A study of sotatercept in participants with PAH WHO FC III or FC IV at high risk of mortality (MK-7962-006/ZENITH) (ZENITH). ClinicalTrials.gov. Updated March 7, 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT04896008
- A long-term follow-up study of sotatercept for PAH treatment (MK-7962-004) (SOTERIA). ClinicalTrials.gov. Updated March 15, 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT04796337
- Andre P, Joshi SR, Briscoe SD, Alexander MJ, Li G, Kumar R. Therapeutic approaches for treating pulmonary arterial hypertension by correcting imbalanced TGF-β superfamily signaling. Front Med (Lausanne). 2022;8:814222. doi.org/10.3389/fmed.2021.814222
- Humbert M, McLaughlin V, Gibbs JSR, et al; PULSAR Trial Investigators. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. 2021;384(13):1204-1215. doi:10.1056/NEJMoa2024277
- Humbert M, McLaughlin V, Gibbs JSR, et al; PULSAR Trial Investigators. Sotatercept for the treatment of pulmonary arterial hypertension. Supplementary Appendix. N Engl J Med. 2021;384(13):1204-1215. doi:10.1056/NEJMoa2024277
- Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J. 2023;61(1):2201347. doi:10.1183/13993003.01347-2022
- Torres F, Farber H, Ristic A, et al. Efficacy and safety of ralinepag, a novel oral IP agonist, in PAH patients on mono or dual background therapy: results from a phase 2 randomised, parallel group, placebo-controlled trial. Eur Respir J. 2019;54(4):1901030. doi:10.1183/13993003.01030-2019
- Pitre T, Su J, Cui R, et al. Medications for the treatment of pulmonary arterial hypertension: a systematic review and network meta-analysis. Eur Resp Rev. 2022;31:220036. doi:10.1183/16000617.0036-2022
- Chang SA, Kim HK, Chang HJ, Kim DK. Acute hemodynamic changes after single administration of udenafil in pulmonary arterial hypertension: a phase IIa study. Korean Circ J. 2019;49(4):353-360. doi:10.4070/kcj.2018.0281
- Chang HJ, Song S, Change SA, et al. Efficacy and safety of udenafil for the treatment of pulmonary arterial hypertension: a placebo-controlled, double-blind, phase IIb clinical trial. Clin Ther. 2019;41(8):1499-1507. doi:10.1016/j.clinthera.2019.05.006
- Pulmonary arterial hypertension. Northern Therapeutics. Accessed February 24, 2023. https://www.northernther.com/pages/2/index.htm

