
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Antineutrophil Cytoplasmic Antibody (ANCA) Vasculitis: Pathophysiology, Diagnosis, and the Evolving Treatment Landscape
Abstract
The antineutrophil cytoplasmic antibody (ANCA) vasculitides include several closely related, often severe, multisystem autoimmune diseases characterized by antibodies against serine proteinase 3 or myeloperoxidase. Loss of tolerance to these antigens triggers a cascade of events, beginning with the priming of neutrophils by proinflammatory cytokines and complement activation, translocation of ANCA-specific antigens to the plasma membrane, neutrophil hyperactivation, and further activation of the alternative complement pathway, leading to tissue damage and the clinical manifestations of ANCA vasculitis. Due to the heterogeneity in presentation of these diseases, diagnosis is often substantially delayed, leading to poor outcomes. The current treatment pathway for most patients involves induction with cyclophosphamide or rituximab in combination with glucocorticoids, followed by a maintenance phase with rituximab, azathioprine, or methotrexate, during which time glucocorticoids are tapered. Current therapies are often effective in inducing and maintaining remission but are associated with a range of toxicities. Several new therapies are in development for ANCA vasculitis. Avacopan, an orally administered inhibitor of the complement fragment 5a (C5a) receptor, has been assessed in a phase 3 clinical trial and may play a role in reducing the cumulative glucocorticoid dose. Preliminary data suggest that cluster of differentiation (CD) 80 and CD86 blockade with abatacept may also have a role in the management of ANCA vasculitis. There is an unmet need for additional therapeutic options for patients with these diseases.
Am J Manag Care. 2021;27(suppl 15):S267-S276. https://doi.org/10.37765/ajmc.2021.88746
Introduction
The antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (eg, ANCA vasculitis) are a heterogeneous group of rare, life-threatening, immune-mediated disorders that are characterized by a necrotizing inflammatory process predominantly affecting small- and medium-sized blood vessels (eg, arterioles, capillaries, and venules).1 There are 3 subtypes of ANCA vasculitis: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). GPA and EGPA are associated with the formation of necrotizing granulomatous inflammation in small to medium vessels; MPA is associated with necrotizing vasculitis primarily affecting small vessels.1 Blood vessel rupture or occlusion results in a broad range of organ-threatening and often life-threatening clinical manifestations.2
Prompt diagnosis of ANCA vasculitis is critical because the organ damage may be life-threatening and irreversible. Symptoms can mimic other diseases (most commonly pneumonia and certain malignancies), leading to misdiagnosis and significant diagnostic delays. More than one-third of patients may not receive a diagnosis for at least 6 months, while nearly a quarter remain undiagnosed for over a year after symptom onset.2,3 The consequences of late diagnosis of ANCA vasculitis are severe, with only about 1 in 10 untreated patients surviving for more than 2 years.4 In contrast, prompt diagnosis and aggressive treatment is associated with dramatically improved outcomes, with remission occurring in 60% to 90% of patients and 2-year survival rates exceeding 90%.2
The current standard of care for ANCA vasculitis includes 2 phases: an initial phase aimed at achieving remission and a longer-term maintenance phase. Remission induction involves high-dose glucocorticoids in combination with cyclophosphamide or rituximab. The subsequent maintenance phase usually involves rituximab, methotrexate, azathioprine, or mycophenolate mofetil, sometimes in combination with reduced-dose glucocorticoids.5 Current treatments for ANCA vasculitis have a substantial degree of efficacy in inducing and maintaining remission in many patients; however, at the dosages and durations of therapy used in ANCA vasculitis, glucocorticoid adverse effects (AEs), such as infection, osteoporosis, diabetes, hypertension, and psychological AEs, among others, can be problematic. Furthermore, cyclophosphamide is associated with a range of well-recognized AEs. These treatment-related AEs, rather than active vasculitis, are the most common cause of death during the first year of ANCA vasculitis treatment.6 There is a significant clinical need for more effective and better-tolerated regimens for these diseases.
This review provides an overview of the classification, epidemiology, and pathophysiology of ANCA vasculitis, followed by a discussion of the current management paradigm and potential new therapies.
Classification and Epidemiology of ANCA Vasculitis
The Chapel Hill Consensus Conference defines ANCA vasculitis as a necrotizing vasculitis with few or no immune deposits that predominantly affects small vessels (ie, capillaries, venules, arterioles, and small arteries). It is associated with antibodies against myeloperoxidase (MPO) or serine proteinase 3 (PR3) in most patients.1 The major clinicopathologic variants of ANCA vasculitis include GPA (formerly Wegener granulomatosis [WG]), MPA, and EGPA (formerly Churg-Strauss syndrome).1 The incidence, prevalence, definitions, and features of these diseases are summarized in the Table.7 Considerable pathophysiologic and clinical overlaps are present in GPA and MPA, and they are often studied together in clinical trials.7


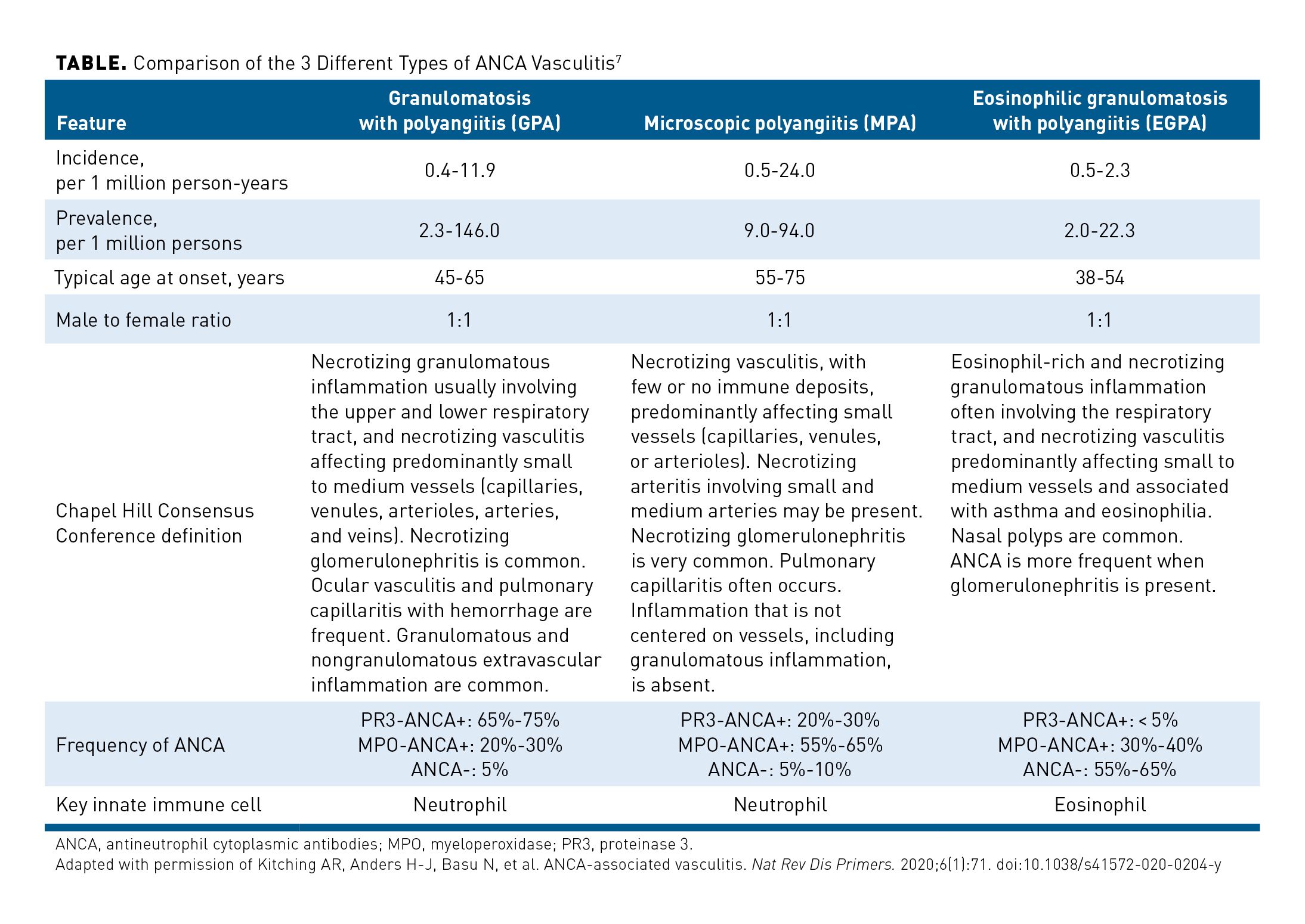
Most patients with ANCA vasculitis test positive for ANCAs during their lifetime, although ANCA-negative cases are seen and have similar manifestations.7,8 PR3-ANCA is most commonly seen in patients with GPA (65%-75%), whereas MPO-ANCA is more common in patients with MPA (55%-65%) and EGPA (30%-40%).7 In clinical practice, patients are typically categorized by clinicopathologic variant, although controversy exists as to whether ANCA serotype might be a more appropriate way to classify patients.9
Pathophysiology of ANCA Vasculitis
ANCA vasculitis is characterized by a loss of tolerance to the neutrophil primary granule protein PR3 (a protease involved in the degradation of extracellular proteins) or MPO (a heme-containing peroxidase that catalyzes the formation of reactive oxygen intermediates involved in microbial killing by neutrophils). Normally, MPO and PR3 are tightly sheltered from the immune system and are quickly neutralized after degranulation to limit the development of autoantibodies.7,10-13 Proposed mechanisms underlying the loss of tolerance in ANCA vasculitis and the initiation of autoantibody production have been described elsewhere.7,12,13
Neutrophil priming is initiated when an activating stimulus consisting of a combination of factors, including inflammatory cytokines, such as tumor necrosis factor-α, interleukin (IL)-1, and IL-18 results in the delocalization of MPO and PR3 to the surface of the neutrophils, allowing ANCA autoantibodies to bind (Figure 1).14 Priming leads to the generation of complement fragment 5a (C5a), a major downstream effector molecule in the alternative complement pathway. Further neutrophil priming occurs when C5a binds to the C5a receptor (C5aR), activating neutrophils and attracting more neutrophils to the inflammatory site, and the subsequent production of neutrophil extracellular traps (NETs).12,13,15-17


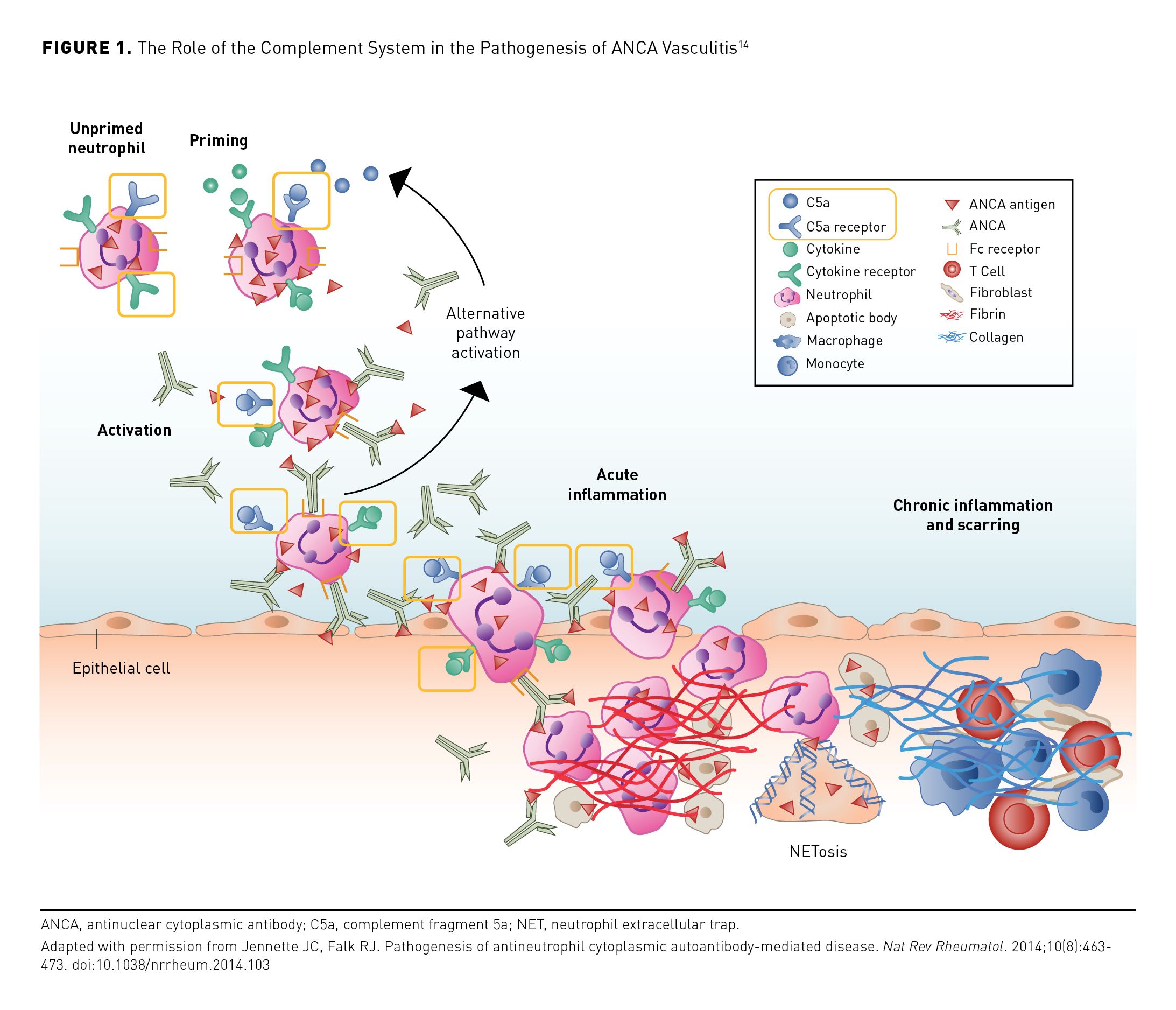
The presence of necrotizing granulomatous inflammation in the respiratory tract is a key histopathologic feature of GPA that is not seen in MPA. Granulomatous inflammation is thought to be initiated by infection, such as that by Staphylococcus aureus. The resulting activation of macrophages in the respiratory epithelia recruits neutrophils and monocytes to the area of inflammation. Recruited neutrophils encounter the bacteria and release reactive oxygen species and undergo lysis, forming the necrotic center of the lesion. Monocytes recruited to the site differentiate into IL-23–secreting macrophages, which drive local T cells toward a T helper 17 (Th17) phenotype. The Th17 cells released from these cells elicit the development of the granulomatous lesions that surround the necrotic core of the lesion.12
Clinical Features and Diagnosis of ANCA Vasculitis
Clinical Features
The clinical features of ANCA vasculitis are heterogeneous and depend on the variant, organs involved, and extent of damage. ANCA vasculitis may affect any organ, but it is most commonly seen in the lungs; the kidneys are also frequently involved.2
ANCA vasculitis may present with constitutional symptoms and symptoms associated with specific organ involvement. Constitutional symptoms vary but include myalgias, loss of appetite, weight loss, fever, night sweats, and migratory arthropathy.2,19 Some patients may present with prodromal symptoms that occur weeks to months prior to the onset of clinically apparent end-organ involvement.19
Involvement of the upper and lower respiratory tract is observed in ANCA vasculitis, but presentation varies between the different subtypes.7,19 Pulmonary involvement is seen in 90% of patients with GPA and at least 70% of those with EGPA; it is less frequently seen in patients with MPA (50%).19 It commonly co-occurs in patients with glomerulonephritis. The severity of pulmonary involvement may range from a transient infiltration, most commonly seen in EGPA, to cavitary lung nodules observed in GPA. Bronchial inflammation may be seen in GPA and EGPA, while hemorrhage can occur in all 3 diseases. Patients may present with hemoptysis, shortness of breath, and cough. Patients with EGPA universally have asthma.2
Involvement of the upper airway is observed in up to 90% of patients with GPA; it is less common in patients with MPA (35%) and EGPA (50%).19 Sinonasal disease may be characterized by chronic sinusitis, nasal crusting, and epistaxis. The inflammation in GPA can result in tissue destruction causing nasal septal perforation and saddle nose deformity. Paranasal sinus disease and nasal polyps are more common in EGPA. Sinusitis may be the presenting feature in many patients until clinically apparent multiorgan disease occurs.8,20
Kidney disease is one of the most clinically morbid consequences of ANCA vasculitis. The prevalence of renal involvement varies according to subtype, occurring in about 90% of patients with MPA, 80% of those with GPA, and 45% of those with EGPA.19 Manifestations may include hematuria, proteinuria, and reduced glomerular filtration rate (GFR); if left untreated, renal inflammation can result in rapid evolution to end-stage renal disease (ESRD) requiring renal replacement therapy or transplant.6,21
As compared to those without ANCA vasculitis, patients with ANCA vasculitis have a 3-fold increased risk for cardiovascular disease (CVD), an 8-fold increased risk for cerebrovascular accidents, and a 6-fold increased risk for venous thromboembolism.22,23 The increased risk of CVD is seen in the absence of risk factors, including elevated low-density lipoprotein cholesterol, depressed high-density lipoprotein cholesterol, smoking, or other cardiovascular risk factors as measured by the Framingham Risk Score.22 ANCA vasculitis is known to promote a hypercoagulable state that is poorly understood; however, it may be the result of the release of microparticles and NETs-carrying tissue factor.16
Peripheral neuropathy is most common in EGPA (70%), followed by GPA (50%) and MPA (30%). It is caused by vasculitis of the small arteries that supply the peripheral nerves and may present as sensory neuropathy or mononeuritis multiplex.7,19,24
Other organ systems may also be affected in ANCA vasculitis. Skin involvement is common and usually presents as purpura in the lower extremities, although other cutaneous manifestations such as nodules, petechiae, ecchymoses, and ulcers may occur.19,25 Gastrointestinal involvement may present as abdominal pain or hematochezia in at least half of patients with ANCA vasculitis. Intestinal infiltration with eosinophils may cause abdominal pain in patients with EGPA.26 Ocular symptoms, such as eye pain due to scleritis or diplopia as a result of a retro-orbital mass, develop in 20% of patients and are seen more frequently in GPA.2,27
Diagnosis of ANCA Vasculitis
The diagnosis of ANCA vasculitis is made clinically, with support from serologic and histologic findings.2 Although many of the signs and symptoms discussed above can be seen in other disease states, multisystem involvement is a key diagnostic clue, and the index of suspicion should be high if at least 2 of these symptoms are observed.2 An ANCA test should be performed in any adult who presents with symptoms clinically suggestive of ANCA vasculitis, but a negative test does not exclude the disease.2 Imaging showing destructive sinus disease, lung nodules, or opacities can assist in the diagnosis. Biopsy of affected organs is recommended to help establish a diagnosis of ANCA vasculitis.5 Kidney and lung biopsies offer the highest yield for supporting a diagnosis.5
Current Treatment of ANCA Vasculitis
Treatment of severe ANCA vasculitis is usually divided into 2 phases. The first is a remission-induction phase, which is an initial, short phase whereby patients are treated with high-dose glucocorticoids in combination with cyclophosphamide or rituximab, sometimes in combination with other agents, to abate inflammation. This is followed by a remission maintenance phase using lower doses of glucocorticoids, often in combination with a medication that has fewer potential AEs and can be used for longer periods with less frequent toxicity monitoring.5
Current treatments for ANCA vasculitis are effective but can be associated with a range of severe and sometimes fatal AEs. This is particularly true for cyclophosphamide, one of the first agents with clear clinical evidence for induction of remission and reduced mortality in patients with ANCA vasculitis. Although effective, AEs such as hemorrhagic cystitis, ovarian and testicular failure, anemia, thrombocytopenia, and urologic and hematologic malignancies have led health care providers to find ways to reduce or eliminate patient exposure to this agent.
Most recent large, controlled studies in ANCA vasculitis have evaluated the treatment of GPA and MPA; for this reason, the management of EGPA with conventional synthetic agents has largely been inferred from the results of these studies. A single agent, mepolizumab, has demonstrated efficacy in a clinical trial focused on EGPA and is currently indicated for the treatment of these patients.5,7
Remission Induction
The combination of glucocorticoids and cyclophosphamide or rituximab is considered the current standard for induction of remission in most patients with severe disease manifestations, such as rapidly progressive glomerulonephritis, central nervous system vasculitis, cardiac involvement, ischemic bowel disease, scleritis, or severe peripheral neuropathy.5 Rituximab may be preferred in relapsing patients who have PR3-ANCA, and it is often favored in younger patients who plan to have children.5,28 In some patients, methotrexate and mycophenolate mofetil are options for remission induction.5
Glucocorticoids have robust and rapid anti-inflammatory effects. Intravenous (IV) methylprednisolone has conventionally been administered at high doses of 500 to 1000 mg daily for 3 days followed by conversion to oral prednisone 1 mg/kg/d for up to 4 weeks in patients with critical organ involvement. This regimen is followed by a slow taper over several months, with a goal of complete discontinuation or maintenance of the medication at doses of 5 mg/d or less in patients who continue to be in remission. For example, in the RAVE study, conducted by Stone and colleagues, patients were treated with 1 to 3 pulses of methylprednisolone followed by prednisone 1 mg/kg/d. The dose of prednisone was subsequently tapered so that all patients who remitted without flares discontinued glucocorticoids after 5 months.28
Glucocorticoids are associated with a broad range of well-recognized AEs, including increased risk for infection, osteoporosis, diabetes, hypertension, psychosis, insomnia, skin thinning, early cataracts, glaucoma, bruising, and striae.8 Regimens that reduce the amount of glucocorticoid exposure are highly desirable. In the PEXIVAS trial, Walsh and colleagues compared a standard-dose glucocorticoid regimen with a reduced-dose regimen.29 Both groups were treated with a weight-based oral prednisone or prednisolone regimen at the beginning of the study. The dose was reduced by 50% in the reduced-dose group at the start of the second week, whereas the dose in the standard-dose group was tapered more gradually, beginning in the third week. All patients in both groups were given 5 mg/d of prednisone or prednisolone beginning at week 23. Both groups received cyclophosphamide or rituximab and plasma exchange or no plasma exchange. This strategy resulted in a cumulative dose of glucocorticoids that was 60% lower among patients in the reduced-dose group compared with the standard-dose group. The reduced dosing strategy was noninferior to standard dosing and was associated with a 31% reduction in serious infections after the first year. However, the overall types and frequencies of serious AEs were generally similar in the 2 groups, although serious adverse kidney-related events were more common in the reduced-dose group (risk ratio, 1.84; 95% CI, 1.18-2.87).
The combination of glucocorticoids and cyclophosphamide has been used for remission induction for over 4 decades. In one of the earliest studies of cyclophosphamide in this setting, Fauci and colleagues treated patients with cyclophosphamide 2 mg/kg/d plus prednisone 1 mg/kg/d for 2 to 4 weeks, followed by conversion of the glucocorticoid component to alternate days over 1 to 2 months. Ninety-three percent of patients achieved a complete remission that lasted for a mean of 48 months.4 Similarly, Jayne and colleagues showed that the combination of oral daily cyclophosphamide (2 mg/kg/d) with prednisone (starting at 1 mg/kg/d with tapering to 0.25 mg/kg/d by 12 weeks) for 3 to 6 months induced remission in 93% of patients; however, severe or life-threatening AEs occurred in 10% of patients.30 The subsequent CYCLOPS trial, conducted by de Groot and colleagues, compared a pulsed regimen of IV cyclophosphamide (10-15 mg/kg every 2-3 weeks) with oral cyclophosphamide 2 mg/kg/d that resulted in a lower cumulative dose of the drug. This regimen was as effective as oral daily cyclophosphamide for remission induction and was associated with fewer AEs; however, a retrospective follow-up study found that patients who received the daily oral regimen had a lower risk for relapse over a median of 4.3 years compared with those who received the pulsed IV regimen.31,32
At the dosages and durations used in conventional remission induction regimens, the combination of glucocorticoids with cyclophosphamide is associated with considerable toxicities, resulting in a range of serious AEs and even death. In 1 study of 524 patients with newly diagnosed ANCA vasculitis who received this combination, the 1-year probability of death was 11.1%, with 59% of these deaths attributed to AEs associated with the medications rather than active vasculitis (14%).6
Because of its favorable safety profile and proven efficacy, rituximab may be preferred as an induction agent in many patients.5,7 The RAVE study compared the combination of glucocorticoids with rituximab or cyclophosphamide for remission induction in 197 patients with GPA or MPA, of whom approximately 48% had newly diagnosed disease.28 The primary end point was disease remission without the use of prednisone for 6 months, with remission defined as a score of 0 for the Birmingham Vasculitis Activity Score (BVAS) for WG (score range, 0 to 63; higher scores indicate more active disease). In this study, rituximab was noninferior to cyclophosphamide for the primary end point, which was met in 64% of patients given rituximab and 53% given cyclophosphamide.
Among patients with relapsed disease, remission rates were significantly higher with rituximab than with cyclophosphamide (67% vs 42%; P = .01). The rate of AEs was similar in the 2 groups.33
Plasma exchange was thought to be a rapid way to remove pathogenic ANCAs and other proinflammatory proteins involved in the pathogenesis of ANCA vasculitis. Past studies have suggested that it may improve survival for participants with diffuse alveolar hemorrhage and renal failure and may also reduce progression to ESRD compared with IV methylprednisolone.5,34 The PEXIVAS trial (described previously) also randomized patients to undergo plasma exchange (7 plasma exchanges within 14 days after randomization) or no plasma exchange.29 There was no evidence for the efficacy of plasma exchange; in this trial, death or ESRD (the composite primary end point) occurred in 28.4% of patients in the plasma exchange group and 31.0% of those in the control group (HR, 0.86; 95% CI, 0.65-1.13; P = .27). However, patients with pulmonary hemorrhage may have been underrepresented in the study.
Among patients with nonsevere disease, alternative immunosuppressive agents may be used for remission induction.5 Methotrexate (administered orally or parenterally) and mycophenolate mofetil, both in combination with glucocorticoids, have demonstrated equivalent efficacy to cyclophosphamide-based regimens for remission induction in this setting.7,35,36 However, long-term follow-up suggests that patients managed initially with methotrexate have less effective disease control compared with patients who receive cyclophosphamide.37 For this reason, methotrexate is not usually used as an induction agent in patients with severe, organ-threatening disease.5 Mycophenolate mofetil is associated with a higher relapse rate than cyclophosphamide in PR3-positive patients but not MPO-positive patients.36
Remission Maintenance
Once remission has been achieved, long-term maintenance therapy with azathioprine, rituximab, methotrexate, or mycophenolate mofetil, sometimes in combination with low-dose glucocorticoids, may be recommended. The glucocorticoid dose should be tapered to the lowest effective dose.5 Use of cyclophosphamide for more than 3 to 6 months is generally not recommended due to potential toxicities associated with this treatment that limit its applicability for longer-term therapy.5 Azathioprine has been shown to be a more effective maintenance agent compared with mycophenolate mofetil.38
Rituximab given at 500 mg every 6 months or 1000 mg every 4 months has consistently shown lower relapse rates compared with azathioprine. A recent expert consensus guideline recommends rituximab for remission maintenance in patients who receive a rituximab-based regimen for remission induction. They also suggest that rituximab be considered for maintenance in patients who received a cyclophosphamide-based regimen for remission induction.39
These recommendations were based, in part, on results from the MAINRITSAN trial. In this study, patients with newly diagnosed or relapsing GPA or MPA in complete remission after a cyclophosphamide-glucocorticoid regimen were randomly allocated to maintenance therapy with daily oral azathioprine or a fixed-schedule regimen of rituximab (two 500-mg doses of rituximab administered on days 0 and 14 followed by 500 mg every 6 months until month 18).40 Major relapse, the primary end point, occurred in 5% of patients who received rituximab, compared with 29% of those who received azathioprine; further, remission rates remained higher with rituximab than with azathioprine through 60 months of follow-up.41 The MAINRITSAN 2 trial showed that redosing rituximab (500 mg) at the return of CD19-positive B cells or a change in ANCA titers is superior to azathioprine for the maintenance of remission.42 However, it is unclear if fixed dosing of rituximab is superior to that of a tailored dose approach based on CD19 or ANCA changes.
The MAINRITSAN 3 trial evaluated whether prolonged rituximab treatment could prevent relapses in patients with GPA or MPA. Sixty-eight patients who achieved complete remission after the first phase of maintenance therapy were randomized to either rituximab or placebo infusion every 6 months for 18 months (4 infusions). Extended rituximab therapy was associated with a significantly higher rate of relapse-free survival (96%) at month 28 compared with no additional therapy (74%; P = .008).43
Unaddressed Needs in the Treatment of ANCA Vasculitis
Early treatment of ANCA vasculitis can reduce or prevent accumulation of organ damage. However, patients remain at a 9-fold increased risk for death in the first year of treatment compared with healthy matched controls, due to inadequate suppression of disease progression and treatment-related AEs.6,44 Furthermore, up to 38% of patients with treated ANCA vasculitis develop ESRD within 5 years due to inadequately controlled disease.21,45-47
As in many autoimmune diseases, glucocorticoids are highly effective in controlling ANCA vasculitis. However, these agents are, by nature, poor long-term therapies due to a broad range of well-understood limitations, including increased risk for infection, diabetes, bone fragility and fracture, elevated blood pressure, cataracts, and gastrointestinal bleeding. Weight gain is common, as are sleep disturbances, lipodystrophy, and neuropsychiatric disturbances.48,49 The impact of long-term treatment with glucocorticoids was assessed in the European Vasculitis Study Group therapeutic trials. In the 270 patients included in this analysis with data at baseline, 6 months, 12 months, and at long-term follow-up, “treatment-related damage” typical of the long-term AEs of glucocorticoid treatment was seen in the majority of patients, including hypertension (41%), osteoporosis (14.1%), and diabetes (10.4%).50
Advances in the Treatment of ANCA Vasculitis
Current regimens have limitations in terms of efficacy and safety. In particular, glucocorticoid-sparing treatment regimens are a critical unmet need. Several new therapies are in late-stage development for ANCA vasculitis, including abatacept, avacopan, and belimumab. Of these, avacopan, an orally administered inhibitor of the complement C5a receptor, is the only agent that has been assessed in a phase 3 clinical trial.17,51,52 Although limited to results from a small, open-label study, the selective costimulation modulator abatacept has shown promising results.53 Belimumab, a monoclonal antibody directed against B-lymphocyte stimulator (BLyS), has been evaluated, thus far with equivocal results, in combination with azathioprine for the maintenance of remission in patients with GPA and MPA.54 A number of other agents are in early stages of development and are not addressed here.
Abatacept
Abatacept is a soluble fusion protein that consists of the extracellular domain of the human cytotoxic T-lymphocyte-associated antigen 4 linked to a modified Fc portion of human immunoglobulin G. It acts by binding to cluster of differentiation (CD) 80 and CD86, blocking this critical costimulatory signal that is necessary for the full activation of T lymphocytes.55
Abatacept was evaluated in a small (N = 20) open-label, prospective trial in patients with a nonsevere relapse of GPA.53 Patients were treated with 3 doses of weight-based intravenously administered abatacept during the first month, followed by monthly infusions for a minimum of 6 months. Patients were allowed to receive up to 30 mg/d of prednisone at study entry, which was subsequently tapered to the lowest extent possible. Patients who received methotrexate, azathioprine, or mycophenolate mofetil could continue treatment with these agents as long as the dose was not increased. The primary end point was disease improvement, as measured by the BVAS for WG.
Among the 20 patients enrolled in the study, disease improvement occurred in 90% and remission (a BVAS score of 0) was achieved in 80% at a median of 1.9 months.53 The median duration of remission before the closing of the trial was 14.4 months. Eleven patients of the 15 who were taking prednisone (73%) tapered completely off glucocorticoids. A total of 6 patients (30%) experienced increased disease activity and thus met criteria for early termination. In total, there were 9 serious AEs, 7 of which were infections, all of which were treated without further incident.
A phase 3 clinical trial of abatacept for the treatment of relapsing non-severe GPA is ongoing and expected to enroll approximately 66 patients (NCT02108860).56 Patients are maintained on a stable dose of their maintenance immunosuppressive agent (methotrexate, azathioprine, or mycophenolate mofetil) and are randomized to abatacept or placebo plus a tapering regimen of prednisone starting at 30 mg daily.
Avacopan
The alternative complement pathway, as discussed previously, is an integral driver of ANCA vasculitis pathogenesis. Avacopan is an orally administered small molecule C5a receptor antagonist that selectively blocks the effects of C5a through C5aR. In a transgenic mouse model, avacopan was highly effective in reducing anti-MPO antibody-induced glomerulonephritis and was a potent inhibitor of C5a-induced neutrophil migration and margination in cynomolgus monkeys.52,57
Phase 2 clinical trials (CLEAR and CLASSIC) provided initial evidence that avacopan had efficacy in ANCA vasculitis.54,58 In the CLEAR trial, 67 patients with GPA or MPA were randomized to avacopan plus placebo, avacopan with low-dose prednisone (20 mg/d with taper), or placebo plus standard-dose prednisone (60 mg/d), all in combination with cyclophosphamide followed by azathioprine or rituximab.54 Clinical responses occurred in 70% of the standard treatment group, 86% of the avacopan plus reduced-dose prednisone group (P = .002 for noninferiority vs standard care), and 81% of the avacopan without prednisone group (P = .01 for noninferiority vs standard of care). A rapid improvement in proteinuria was observed among patients who received avacopan. Avacopan was also associated with fewer glucocorticoid-associated adverse effects and improvements in health-related quality of life. The CLASSIC study, which enrolled 42 patients with newly diagnosed or relapsing ANCA vasculitis, randomized subjects to 12 weeks of treatment with either twice daily avacopan 10 mg or 30 mg plus standard of care or standard of care alone.58 In this small study, there was little difference in the rate of serious AEs between the 2 groups (15% for standard of care and 17% for avacopan plus standard of care). Response on the BVAS was similar and high across arms; however, increases in the mean vascular damage index were greater with standard of care than with avacopan plus standard of care.
Avacopan was further evaluated in the phase 3 ADVOCATE trial, a multicenter, international, randomized, double-blind, double-dummy trial.51 Patients enrolled in the trial had newly diagnosed or relapsing GPA or MPA that qualified for treatment with cyclophosphamide or rituximab. Enrolled patients tested positive for antibodies to PR3 or MPO, had an estimated GFR of greater than or equal to 15 mL/min/1.73 m2, and had 1 or more major items, 3 nonmajor items, or 2 or more renal items for the BVAS. Patients were required to cease immunosuppressants before trial entry.
Patients were randomly allocated to oral treatment with 30 mg of avacopan twice daily or a tapering regimen of prednisone.51 All patients received concomitant cyclophosphamide (followed by azathioprine) or rituximab. Patients with worsening disease could receive rescue therapy with IV glucocorticoids (typically 3 days of methylprednisolone), oral glucocorticoids, or both, which were subsequently tapered as appropriate for the patient’s condition.
The first primary end point was remission (BVAS of 0 at week 26 with no glucocorticoid use during the 4 weeks before week 26); the second primary end point was sustained remission (remission at both weeks 26 and 52 with no glucocorticoid use during the 4 weeks before week 52).51 Glucocorticoid-induced toxic effects as assessed using the Glucocorticoid Toxicity Index (GTI) was a key secondary end point.
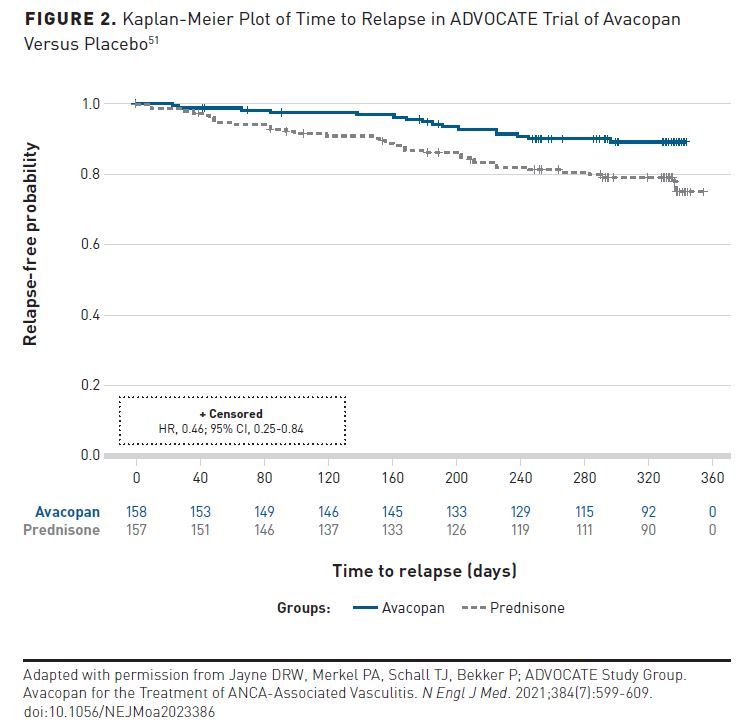
The study enrolled 331 patients, of whom approximately 69% were newly diagnosed and 31% had relapsed disease.51 The first primary end point of remission at week 26 was observed in 72.3% of patients in the avacopan group and 70.1% of the prednisone group, a difference of 3.4 percentage points (95% CI, –6.0 to 12.8; P < .001 for noninferiority; P = .24 for superiority). Sustained remission was observed in 65.7% of patients given avacopan and 54.9% of those given prednisone, a difference of 12.5 percentage points (95% CI, 2.6-22.3; P < .001 for noninferiority; P = .007 for superiority). A total of 16 of 158 patients (10.1%) and 33 of 157 patients (21.0%) in the prednisone group had relapses. The hazard ratio for relapse after remission (avacopan vs prednisone) was 0.46 (95% CI, 0.25-0.84) (Figure 2).51


Avacopan also appeared to have a beneficial effect on renal function relative to prednisone, with an eGFR change from baseline of 7.3 mL/min/1.73 m2 in the avacopan group versus 4.1 mL/min/1.73 m2 in the prednisone group (difference: 3.2 mL/min/1.73 m2; 95% CI, 0.3-6.1).51 Among patients with stage 4 kidney disease, the mean change at week 52 was 13.7 mL/min/1.73 m2 in those given avacopan and 8.2 mL/min/1.73 m2 in those given prednisone (difference: 5.6 mL/min/1.73 m2; 95% CI, 1.7-9.5).
Avacopan was associated with a reduction in the need for glucocorticoids.51 The total mean prednisone-equivalent dose of oral and IV glucocorticoids was 1676 mg (or about 5 mg/patient/d) in the avacopan group and 3847 mg (or about 13 mg/patient/d) in the prednisone group. Scores on the GTI were lower in patients given avacopan compared with those given prednisone (39.7 vs 56.6; difference: –16.8 points; 95% CI, –25.6 to –8.0).
Serious AEs occurred in 42% of patients who received avacopan and 45% of those who received prednisone; the most common serious AE was worsening of vasculitis (10.2% and 14.0% of the 2 groups, respectively).51 There were 2 deaths in the avacopan group (1 each of worsening of vasculitis and pneumonia) and 4 deaths in the prednisone group (2 infections, 1 acute myocardial infarction, and 1 death of unknown cause). Serious infections occurred in 13.3% of patients given avacopan and 15.2% of patients given prednisone. The incidence of AEs potentially related to glucocorticoids was 66.3% in the avacopan group and 80.5% in the prednisone group (difference: –14.2 percentage points; 95% CI, –23.7 to –3.8).
Belimumab
Belimumab, currently indicated for the treatment of systemic lupus erythematosus, is a monoclonal antibody specific for BLyS (also referred to as B-cell activating factor).59 By binding to BLyS, belimumab inhibits the survival of B cells, including autoreactive B cells, and reduces differentiation of B cells into plasma cells.
Belimumab was evaluated by Jayne and colleagues in the BREVAS study. Patients received induction therapy with glucocorticoids plus rituximab or cyclophosphamide for 6 to 26 weeks, followed by randomization to treatment with low-dose glucocorticoids and azathioprine plus belimumab or placebo for maintenance.60 The primary end point was time to first occurrence of a protocol-specified event (PSE), defined as a BVAS of at least 6, presence of at least 1 major BVAS item, or receipt of prohibited medications for any reason, resulting in treatment failure. The addition of belimumab to azathioprine and low-dose glucocorticoids did not reduce the risk for PSEs (HR, 1.07; 95% CI, 0.44-2.59; P = .884); however, for both treatment arms, the rate of such events was only about 20%. No differences were observed in the rate of vasculitis relapse (HR, 0.88; 95% CI, 0.29-2.65; P = .821).
Conclusions
Damage due to active ANCA vasculitis is often cumulative and irreversible. Thus, a prompt diagnosis and early therapy can reduce morbidity or even be lifesaving. Despite the development of new medications that have improved the rate of death, morbidity and mortality remain high due to inadequate suppression of disease progression and treatment-related toxicities.
Current therapeutic strategies for ANCA vasculitis are aimed at reducing exposure to, or eliminating the need for, cyclophosphamide and at reducing the cumulative dose of glucocorticoids. Rituximab represented a major advance in the management of ANCA vasculitis, showing equal efficacy and—in some instances, better efficacy—than cyclophosphamide. However, few studies have demonstrated a consistent reduction in the need for or dosage of glucocorticoids. Thus, there remains a need for additional medications that can allow for reductions in the cumulative steroid dose.
The majority of recent advances in the treatment of ANCA vasculitis have been in the repurposing of therapies originally developed in different disease states and in refining existing therapeutic regimens to improve efficacy and reduce toxicity. These advances have led to substantial improvements in the clinical outlook for these patients, with the increasing use of rituximab as both maintenance and induction therapy being a key example of this approach. Advances in our understanding of ANCA vasculitis and in the development of new therapies for this disease have only recently begun to lead to additional therapeutic approaches. In particular, animal studies have provided useful new insight into the pathogenesis of ANCA vasculitis. These studies show that blocking the interaction of antigen-presenting cells and T cells with the selective costimulation modulator abatacept may benefit patients with ANCA vasculitis. Indeed, a small open-label study suggests that abatacept is effective in ANCA vasculitis, and the results of phase 3 studies are eagerly awaited. Evidence also suggests that the complement system may have a more important role in the pathogenesis of this disease than originally thought. Based on results from a phase 3 study of the novel C5aR inhibitor avacopan, this agent may have a role in eliminating or reducing the dose of steroids when given in combination with rituximab or cyclophosphamide. Outcomes in ANCA vasculitis have been slowly but steadily improving over time. As our understanding of the mechanisms of ANCA vasculitis grows, the identification of additional targets for therapeutic intervention will likely lead to improved outcomes in this at-need patient population.
Author Affiliations: University of Utah and George E. Wahlen Department of Veterans Affairs Medical Center (CLK); ChemoCentryx, Inc (IVH).
Funding Source: This supplement was supported by ChemoCentryx, Inc.
Author Disclosures: Dr Koening has acted as a consultant to ChemoCentryx, Inc. Dr von Hennigs has acted as a consultant or on a paid advisory board for, owns stock in, has institutional conflicts of interest with, and is an employee of ChemoCentryx, Inc.
Authorship Information: Concept and design (CLK); Acquisition of data (IVH); Drafting of the manuscript (CLK, IVH); Critical revision of the manuscript for important intellectual content (CLK, IVH).
Address correspondence to: Curry L. Koening, MD, MS, University of Utah, Division of Rheumatology, c/o Curry Koening, 30 N 1900 E, Salt Lake City, UT 84132. Email: curry.koening@hsc.utah.edu
ERRATUM: Since initial publication in September 2021, a conversion miscalculation was found. In the ADVOCATE trial, which this paper reviews, all glucocorticoids were converted into prednisone-equivalent doses. Recently, a conversion miscalculation was found in the non–study-supplied glucocorticoid dose. There are no changes to the conclusions of the study; however, to be fully transparent the authors suggested correcting the published article. The sentence referring to this study has been revised to, “The total mean prednisone-equivalent dose of oral and IV glucocorticoids was 1676 mg (or about 5 mg/patient/d) in the avacopan group and 3847 mg (or about 13 mg/patient/d) in the prednisone group” to reflect this change. In addition, in Figure 2, the Kaplan-Meier curve lines have been revised to show both curves beginning at 1.0.
REFERENCES
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11. doi:10.1002/art.37715
- Hunter RW, Welsh N, Farrah TE, Gallacher PJ, Dhaun N. ANCA associated vasculitis. BMJ.
2020;369:m1070. doi:10.1136/bmj.m1070 - Pearce FA, Hubbard RB, Grainge MJ, Watts RA, Abhishek A, Lanyon PC. Can granulomatosis with polyangiitis be diagnosed earlier in primary care? a case-control study. QJM. 2018;111(1):39-45. doi:10.1093/qjmed/hcx194
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical
and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983;98(1):76-85.
doi:10.7326/0003-4819-98-1-76 - Yates M, Watts RA, Bajema IM, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. 2016;75(9):1583-1594. doi:10.1136/annrheumdis-2016-209133
- Little MA, Nightingale P, Verburgh CA, et al; European Vasculitis Study (EUVAS) Group. Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis.
Ann Rheum Dis. 2010;69(6):1036-1043. doi:10.1136/ard.2009.109389 - Kitching AR, Anders H-J, Basu N, et al. ANCA-associated vasculitis. Nat Rev Dis Primers. 2020;6(1):71. doi:10.1038/s41572-020-0204-y
- Wallace ZS, Miloslavsky EM. Management of ANCA associated vasculitis. BMJ. 2020;368:m421. doi:10.1136/bmj.m421
- Hilhorst M, van Paassen P, Tervaert JWC; Limburg Renal Registry. Proteinase 3-ANCA vasculitis versus myeloperoxidase-ANCA vasculitis. J Am Soc Nephrol. 2015;26(10):2314-2327. doi:10.1681/ASN.2014090903
- Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47-52. doi:10.1016/j.abb.2018.01.004
- Sugawara S. Immune functions of proteinase 3. Crit Rev Immunol. 2005;25(5):343-360.
doi:10.1615/critrevimmunol.v25.i5.10 - Nakazawa D, Masuda S, Tomaru U, Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol. 2019;15(2):91-101. doi:10.1038/s41584-018-0145-y
- Hutton HL, Holdsworth SR, Kitching AR. ANCA-associated vasculitis: pathogenesis, models,
and preclinical testing. Semin Nephrol. 2017;37(5):418-435. doi:10.1016/j.semnephrol.2017.05.016 - Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. doi:10.1038/nrrheum.2014.103
- Söderberg D, Segelmark M. Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol. 2016;7:256. doi:10.3389/fimmu.2016.00256
- Chen M, Jayne DRW, Zhao M-H. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol. 2017;13(6):359-367. doi:10.1038/nrneph.2017.37
- Trivioli G, Vaglio A. The rise of complement in ANCA-associated vasculitis: from marginal player to target of modern therapy. Clin Exp Immunol. 2020;202(3):403-406. doi:10.1111/cei.13515
- Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity.
J Immunol. 2012;189(6):2689-2695. doi:10.4049/jimmunol.1201719 - Rowaiye OO, Kusztal M, Klinger M. The kidneys and ANCA-associated vasculitis: from pathogenesis to diagnosis. Clin Kidney J. 2015;8(3):343-350. doi:10.1093/ckj/sfv020.
- Yates M, Watts R. ANCA-associated vasculitis. Clin Med (Lond). 2017;17(1):60-64. doi:10.7861/clinmedicine.17-1-60
- Corral-Gudino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, Lerma-Márquez JL. Overall survival, renal survival and relapse in patients with microscopic polyangiitis: a systematic review of current evidence. Rheumatology (Oxford). 2011;50(8):1414-1423. doi:10.1093/rheumatology/ker112
- Berti A, Matteson EL, Crowson CS, Specks U, Cornec D. Risk of cardiovascular disease and venous thromboembolism among patients with incident ANCA-Associated vasculitis: a 20-year population-based cohort study. Mayo Clin Proc. 2018;93(5):597-606. doi:10.1016/j.mayocp.2018.02.010
- Kronbichler A, Leierer J, Gauckler P, Shin JI. Comorbidities in ANCA-associated vasculitis. Rheumatology (Oxford). 2020;59(suppl 3):iii79-iii83. doi:10.1093/rheumatology/kez617
- Suppiah R, Hadden RD, Batra R, et al; European Vasculitis Study Group. Peripheral neuropathy in ANCA-associated vasculitis: outcomes from the European Vasculitis Study Group trials. Rheumatology (Oxford). 2011;50(12):2214-2222. doi:10.1093/rheumatology/ker266
- Micheletti RG, Chiesa Fuxench Z, Craven A, Watts RA, Luqmani RA, Merkel PA; DCVAS Investigators. Cutaneous manifestations of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. 2020;72(10):1741-1747. doi:10.1002/art.41310
- Franco DL, Ruff K, Mertz L, Lam-Himlin DM, Heigh R. Eosinophilic granulomatosis with polyangiitis and diffuse gastrointestinal involvement. Case Rep Gastroenterol. 2014;8(3):329-336. doi:10.1159/000369129
- Schmidt J, Pulido JS, Matteson EL. Ocular manifestations of systemic disease: antineutrophil cytoplasmic antibody-associated vasculitis. Curr Opin Ophthalmol. 2011;22(6):489-495. doi:10.1097/ICU.0b013e32834bdfe2
- Stone JH, Merkel PA, Spiera R, et al; RAVE-ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221-232. doi:10.1056/NEJMoa0909905
- Walsh M, Merkel PA, Peh C-A, et al; PEXIVAS Investigators. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med. 2020;382(7):622-631. doi:10.1056/NEJMoa1803537
- Jayne D, Rasmussen N, Andrassy K, et al; European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349(1):36-44. doi:10.1056/NEJMoa020286
- de Groot K, Harper L, Jayne DRW, et al; EUVAS (European Vasculitis Study Group). Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150(10):670-680. doi:10.7326/0003-4819-150-10-200905190-00004
- Harper L, Morgan MD, Walsh M, et al; EUVAS investigators. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis. 2012;71(6):955-960. doi:10.1136/annrheumdis-2011-200477
- Jones RB, Tervaert JWC, Hauser T, et al; European Vasculitis Study Group. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363(3):211-220. doi:10.1056/NEJMoa0909169
- Jayne DRW, Gaskin G, Rasmussen N, et al; European Vasculitis Study Group. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18(7):2180-2188. doi:10.1681/ASN.2007010090
- de Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52(8):2461-2469. doi:10.1002/art.21142
- Jones RB, Hiemstra TF, Ballarin J, et al; European Vasculitis Study Group (EUVAS). Mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA-associated vasculitis: a randomised, non-inferiority trial. Ann Rheum Dis. 2019;78(3):399-405. doi:10.1136/annrheumdis-2018-214245
- Faurschou M, Westman K, Rasmussen N, et al; European Vasculitis Study Group. Brief Report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64(10):3472-3477. doi:10.1002/art.34547
- Hiemstra TF, Walsh M, Mahr A, et al; European Vasculitis Study Group (EUVAS). Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA. 2010;304(21):2381-2388. doi:10.1001/jama.2010.1658
- Tieu J, Smith R, Basu N, et al. Rituximab for maintenance of remission in ANCA-associated vasculitis: expert consensus guidelines. Rheumatology (Oxford). 2020;59(4):e24-e32. doi:10.1093/rheumatology/kez640
- Guillevin L, Pagnoux C, Karras A, et al; French Vasculitis Study Group. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771-1780. doi:10.1056/NEJMoa1404231
- Terrier B, Pagnoux C, Perrodeau E, et al; French Vasculitis Study Group. Long-term efficacy of remission-maintenance regimens for ANCA-associated vasculitides. Ann Rheum Dis. 2018;77(8):1150-1156. doi:10.1136/annrheumdis-2017-212768
- Charles P, Terrier B, Perrodeau E, et al; French Vasculitis Study Group. Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann Rheum Dis. 2018;77(8):1143-1149. doi:10.1136/annrheumdis-2017-212878
- Charles P, Perrodeau E, Samson M, et al. Long-term rituximab use to maintain remission of antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2020;173(3):179-187. doi:10.7326/M19-3827
- Luqmani R, Suppiah R, Edwards CJ, et al. Mortality in Wegener’s granulomatosis: a bimodal pattern. Rheumatology (Oxford). 2011;50(4):697-702. doi:10.1093/rheumatology/keq351
- Koldingsnes W, Nossent H. Predictors of survival and organ damage in Wegener’s granulomatosis. Rheumatology (Oxford). 2002;41(5):572-581. doi:10.1093/rheumatology/41.5.572
- Takala JH, Kautiainen H, Finne P, Leirisalo-Repo M. Wegener’s granulomatosis in Finland in 1981-2000: risk of dialysis-dependent renal disease. Scand J Rheumatol. 2011;40(4):283-288.
doi:10.3109/03009742.2010.533693 - Mohammad AJ, Segelmark M. A population-based study showing better renal prognosis for proteinase 3 antineutrophil cytoplasmic antibody (ANCA)-associated nephritis versus myeloperoxidase ANCA-associated nephritis. J Rheumatol. 2014;41(7):1366-1373. doi:10.3899/jrheum.131038
- McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. 2008;20(2):131-137. doi:10.1097/BOR.0b013e3282f51031
- Fardet L, Flahault A, Kettaneh A, et al. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Br J Dermatol. 2007;157(1):142-148. doi:10.1111/j.1365-2133.2007.07950.x
- Robson J, Doll H, Suppiah R, et al. Damage in the anca-associated vasculitides: long-term data from the European vasculitis study group (EUVAS) therapeutic trials. Ann Rheum Dis. 2015;74(1):177-184. doi:10.1136/annrheumdis-2013-203927
- Jayne DRW, Merkel PA, Schall TJ, Bekker P; ADVOCATE Study Group. Avacopan for the Treatment of ANCA-Associated Vasculitis. N Engl J Med. 2021;384(7):599-609. doi:10.1056/NEJMoa2023386
- Xiao H, Dairaghi DJ, Powers JP, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol. 2014;25(2):225-231. doi:10.1681/ASN.2013020143
- Langford CA, Monach PA, Specks U, et al; Vasculitis Clinical Research Consortium. An open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener’s). Ann Rheum Dis. 2014;73(7):1376-1379. doi:10.1136/annrheumdis-2013-204164
- Jayne DRW, Bruchfeld AN, Harper L, et al; CLEAR Study Group. Randomized trial of C5a receptor inhibitor Avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017;28(9):2756-2767. doi:10.1681/ASN.2016111179
- Moreland L, Bate G, Kirkpatrick P. Abatacept. Nat Rev Drug Discov. 2006;5(3):185-186. doi:10.1038/nrd1989
- Abatacept for the treatment of relapsing, non-severe, granulomatosis with polyangiitis (Wegener’s). ClinicalTrials.gov. Updated April 14, 2021. Accessed June 7, 2021.
https://www.clinicaltrials.gov/ct2/show/NCT02108860 - Bekker P, Dairaghi D, Seitz L, et al. Characterization of pharmacologic and pharmacokinetic properties of CCX168, a potent and selective orally administered complement 5a receptor inhibitor, based on preclinical evaluation and randomized phase 1 clinical study. PLoS One. 2016;11(10):e0164646. doi:10.1371/journal.pone.0164646
- Merkel PA, Niles J, Jimenez R, et al; CLASSIC Investigators. Adjunctive treatment with avacopan, an oral C5a receptor inhibitor, in patients with antineutrophil cytoplasmic antibody-associated vasculitis. ACR Open Rheumatol. 2020;2(11):662-671. doi:10.1002/acr2.11185
- Dennis GJ. Belimumab: a BLyS-specific inhibitor for the treatment of systemic lupus erythematosus. Clin Pharmacol Ther. 2012;91(1):143-149. doi:10.1038/clpt.2011.290
- Jayne D, Blockmans D, Luqmani R, et al; BREVAS Study Collaborators. Efficacy and safety of belimumab and azathioprine for maintenance of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled study. Arthritis Rheumatol. 2019;71(6):952-963. doi:10.1002/art.40802

