
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Efficacy and Safety of Mavacamten in Adults With Symptomatic Obstructive Hypertrophic Cardiomyopathy: Results of the EXPLORER-HCM Study
This AJMC® Clinical Brief provides key information regarding the EXPLORER-HCM study (NCT03470545), the results of which were originally presented by Iacopo Olivotto, MD, at the European Society of Cardiology Congress 2020 – The Digital Experience, which was held from August 29 to September 1, 2020. The results of EXPLORER-HCM were simultaneously published in The Lancet (doi:10.1016/S0140-6736(20)31792-X). The authors of the publication in The Lancet are Olivotto I, Oreziak A, Barriales-Villa R, et al. Please consult the full publication in The Lancet for complete study information and author affiliations.
BACKGROUND
Cardiomyopathies are defined by abnormalities of the ventricular myocardium that are both structural and functional but that are not explained by either flow-limiting coronary artery disease or abnormal loading conditions.1 Hypertrophic cardiomyopathy (HCM) refers to cardiomyopathy characterized by increased left ventricular (LV) wall thickness that cannot be entirely accounted for by abnormal loading conditions.1 The primary pathophysiologic features of HCM include hypercontractility, disordered relaxation and left ventricular compliance, and impaired cardiac energetics.2 Patients with HCM may experience dynamic ventricular outflow tract (LVOT) obstruction, and obstructive HCM is defined as a peak LVOT gradient of 30 mm Hg or greater.2 Patients with HCM may be highly symptomatic and have impaired functionality and reduced quality of life.3,4 HCM can be progressive, with cardiac remodeling and worsening of cardiac function leading to complications such as atrial fibrillation (AF) or heart failure.1,3 HCM may also lead to sudden cardiac arrest or death.3
β-Blockers are currently recommended as first-line pharmacologic management in patients with symptomatic LVOT obstruction; however, their use is not particularly well studied in the disease.1,5 Additionally, although β-blockers can be effective in reducing symptoms, many patients with HCM are unable to tolerate β-blocker therapy.5 Alternatives to β-blockers include nondihydropyridine calcium channel blockers (such as verapamil and diltiazem) and the antiarrhythmic disopyramide, but all these agents must be used with caution. A decrease in blood pressure with verapamil or diltiazem treatment may cause increased outflow obstruction and consequent pulmonary edema, whereas disopyramide is associated with anticholinergic adverse events (AEs) and prolongation of the QTc interval.1,5 Moreover, none of these pharmaceutical treatment options address the underlying molecular mechanisms of HCM.6
Invasive procedures, most prominently ventricular septal myectomy (Morrow procedure) and percutaneous alcohol septal ablation, are generally more effective than the available drug therapies. However, invasive procedures are reserved for patients with advanced disease (eg, New York Heart Association [NYHA] class III-IV) and are associated with risk of complications.1 The need for surgeons and cardiologists experienced in these procedures to achieve optimal outcomes has also limited the availability of procedures.1,6 Taken together, the limitations associated with current treatment options highlight the need for novel pharmacologic treatments to address obstructive HCM.2,6
Pathophysiological studies have shown that in HCM, dysfunction in the sarcomere results in an excess of actin-myosin cross-bridging. This, in turn, causes excessive energy consumption in addition to hypercontractility and impaired relaxation.2 Some patients with HCM have genetic mutations that are considered pathogenic or likely pathogenic.3 The classification and interpretation of genetic variants have evolved over time, and this is an area of ongoing research.7 In published reports, the percentage of patients with HCM who have disease-causing mutations has ranged from approximately 30% to 45%.6,8
Mavacamten is the first agent in a new class of myosin inhibitors—small molecules that selectively target the underlying pathophysiology of HCM by reducing actin-myosin cross-bridge formation, thus improving excess contractility and utilization of myocardial energy.2 The phase 3 EXPLORER-HCM study was designed to assess the efficacy and safety of mavacamten in patients with symptomatic obstructive HCM.2
METHODS
EXPLORER-HCM was a pivotal phase 3, multicenter, randomized, double-blind study of the safety and efficacy of mavacamten in adults with a clinical diagnosis of obstructive HCM, peak LVOT gradient at least 50 mm Hg, and NYHA class II or III symptoms.2,6 After a screening period of up to 35 days, 251 eligible patients were randomized 1:1 to receive oral mavacamten or placebo for 30 weeks, followed by an 8-week posttreatment period, and after that the option to enter a long-term extension study (MAVA-LTE; NCT03723655).2,6 The starting dose of mavacamten was 5 mg once daily, with the option to adjust the dosage at weeks 8 and 14. During titration, dosing adjustments were guided by mavacamten plasma concentration and echocardiography results. Mavacamten doses of 2.5 mg, 5 mg, 10 mg, or 15 mg were given to reduce the LVOT gradient to less than 30 mm Hg and maintain a mavacamten plasma concentration level within the range of 350 to 700 ng/mL.2,6 Based on the results of pharmacokinetic modeling, 15 mg/day was the highest dose.2 Concomitant use of β-blockers or calcium channel blockers was allowed if they were initiated prior to participation in the study; use of disopyramide was not permitted.2
The primary end point consisted of 2 composite functional end points, and achieving either of these was considered a successful primary outcome. These were defined as changes from baseline of either: (1) an increase of at least 1.5 mL/kg/min in peak oxygen consumption (pVO2), as determined by cardiopulmonary exercise testing in addition to a reduction of at least 1 NYHA functional class, or (2) an increase of at least 3.0 mL/kg/min in pVO2 with no worsening in NYHA functional class.2 Secondary end points consisted of changes from baseline at week 30 in postexercise LVOT gradient, pVO2, NYHA class, and scores on 2 patient-reported outcome (PRO) instruments: the Kansas City Cardiomyopathy Questionnaire–Clinical Summary Score for health-related quality of life and the HCM Symptom Questionnaire Shortness of Breath subscore for HCM core symptoms.2 The primary and secondary end points were designed in collaboration with HCM experts, patients with HCM, and regulatory authorities to comprehensively evaluate outcomes.6
RESULTS
Efficacy
Baseline characteristics were similar between the 2 groups with regard to age, concomitant therapy, NYHA functional class, pVO2, and echocardiographic parameters.6 The starting ejection fraction was 74% in both groups, and the postexercise LVOT gradient was 86 and 84 mm Hg in the mavacamten and placebo groups, respectively. History of AF was less common in the mavacamten group (10%) than in the placebo group (18%), whereas those in the mavacamten group had higher baseline levels of serum N-terminal pro b-type natriuretic peptide (NT-proBNP). Overall, 92% of patients were receiving background β-blockers or calcium channel blockers.6
The results for all primary and secondary end points—which assessed symptoms, exercise capacity and function, LVOT obstruction, and key aspects of quality of life—were highly significant, as shown in Table 1.6 The proportion of patients achieving 1 of the 2 composite primary outcomes was more than double in the mavacamten group versus the placebo group (37% vs 17%; P = .0005). The number of patients achieving both of the primary composite end points was 2.5-fold greater in the mavacamten group (20% vs 8%; P = .0005).6


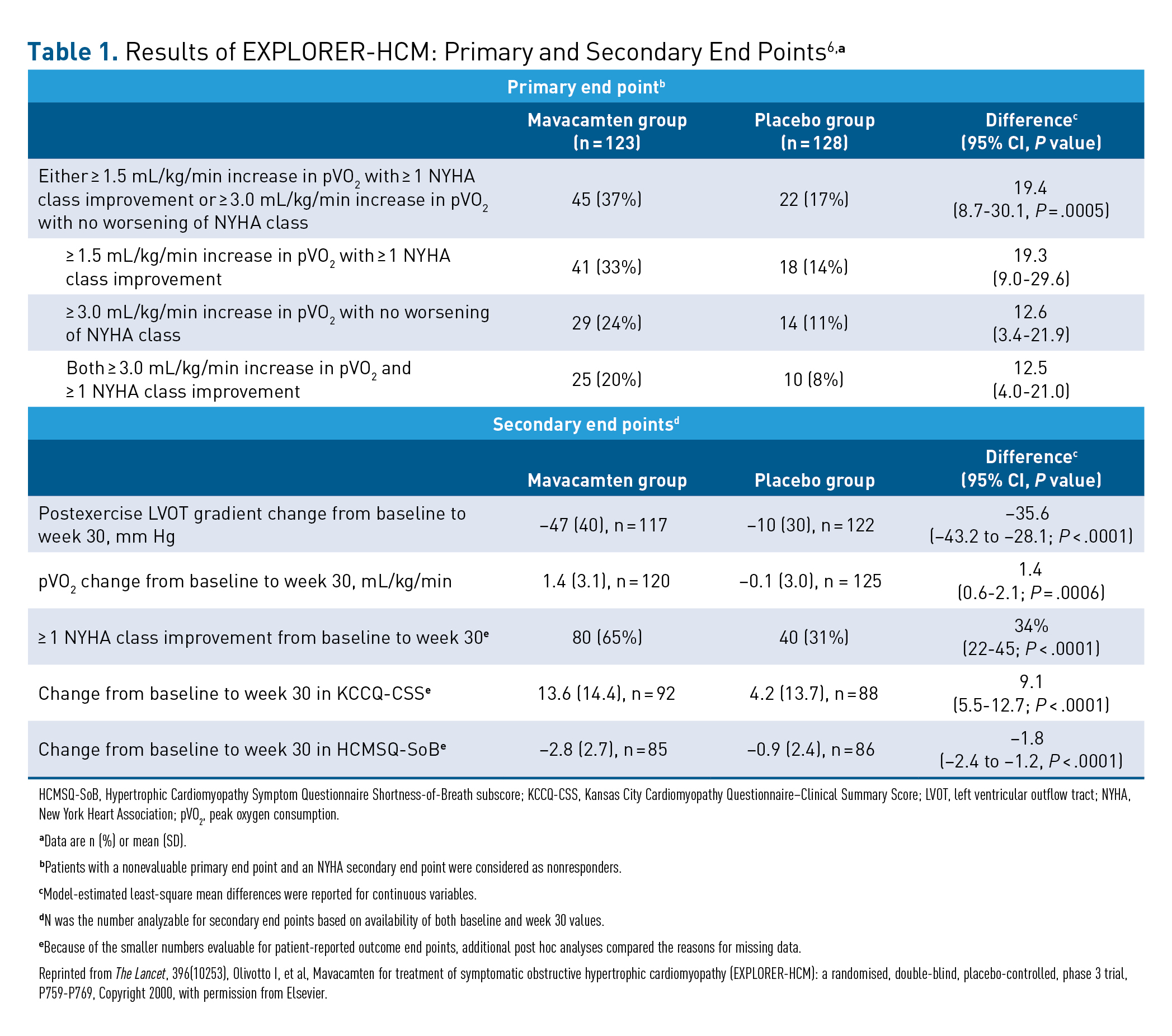
Improvements in the mavacamten group were significantly greater compared with the placebo group for all the secondary end points, including change at week 30 in postexercise LVOT gradient (–47 vs –10; P < .0001) and change in pVO2 (1.4 vs –0.1; P = .0006). Improvement of 1 or more NYHA class was more than twice as common in the mavacamten group (65% vs 31%; P < .0001), and PROs also strongly favored the mavacamten group over the placebo group.6
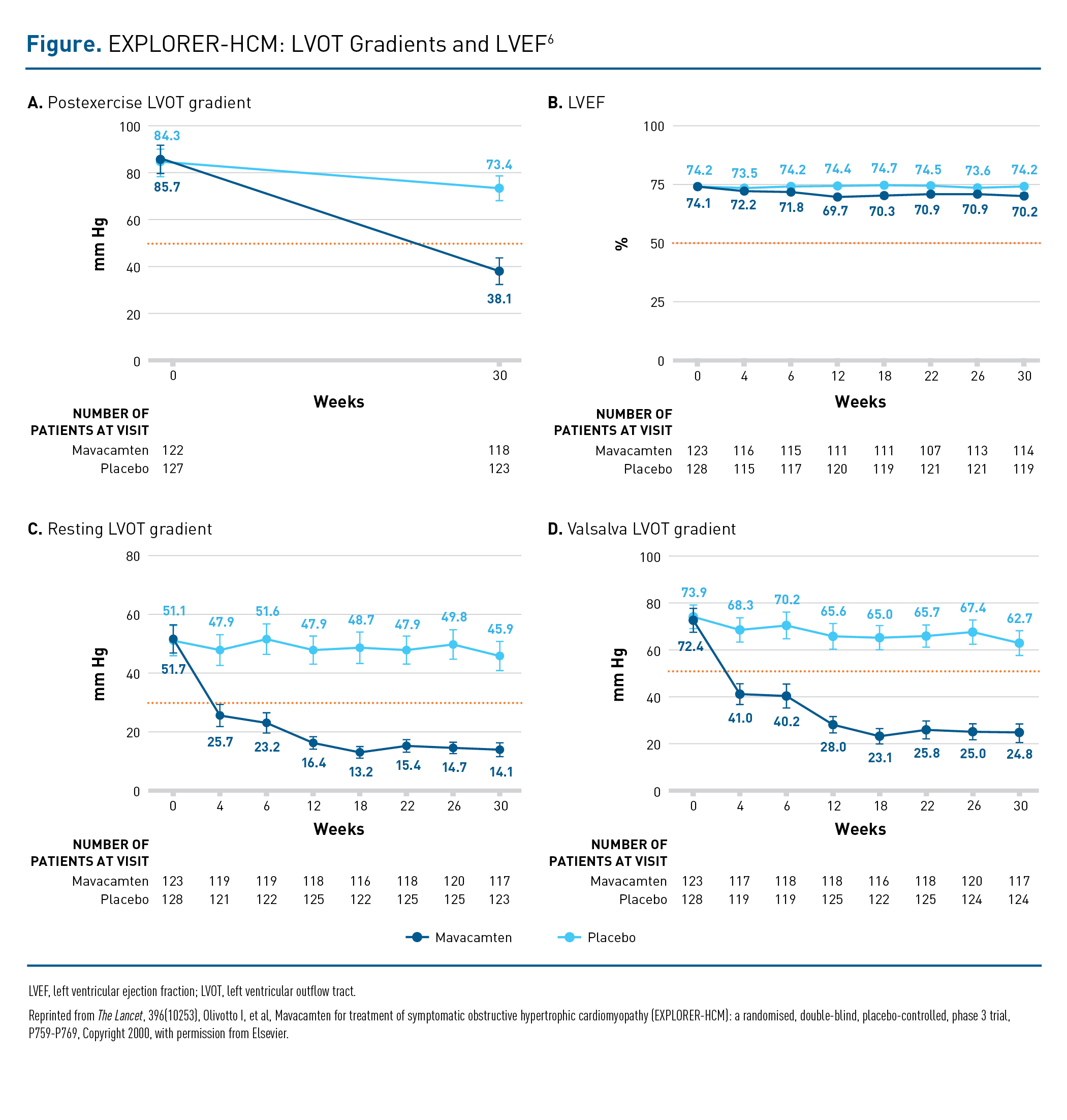
The benefits of mavacamten treatment observed for the secondary end point of postexercise LVOT gradient were seen not just in the overall group but across a variety of patient subgroups, regardless of age, sex, body mass index, baseline left ventricular ejection fraction (LVEF), β-blocker use, baseline NT-proBNP level, and HCM genetic testing results.6 Mavacamten also conferred improvements that occurred early and were sustained in resting and Valsalva LVOT gradients versus placebo (Figure).6


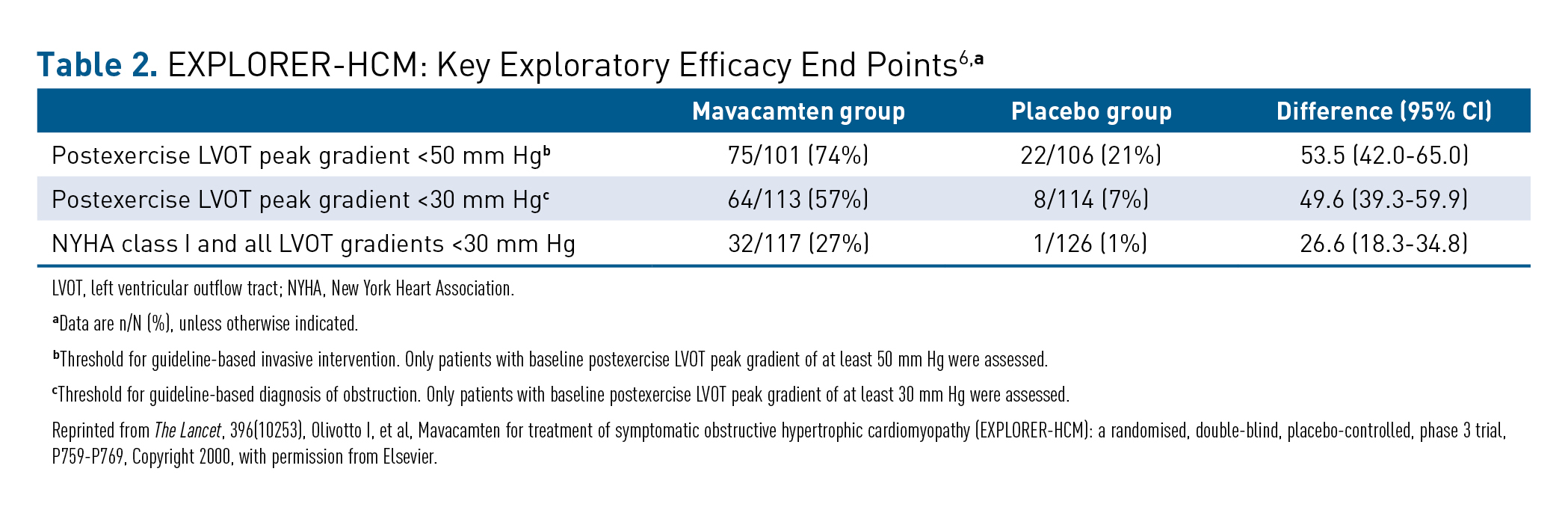
The EXPLORER-HCM trial also assessed several exploratory efficacy end points as shown in Table 2, including the number of patients who achieved a reduction of postexercise LVOT peak gradient to less than 50 mm Hg (ie, below the standard threshold for invasive septal reduction therapy), which was met by 74% versus 21% of the mavacamten and placebo patients, respectively.6 Relief of LVOT obstruction (ie, postexercise gradient < 30 mm Hg) was seen in more than half of all patients treated with mavacamten (57%) compared with 7% in the placebo group.6 In addition, 32 of 117 patients (27%) treated with mavacamten became asymptomatic (defined as being NYHA class I) and were below the guideline-based definition of obstruction (all LVOT gradients < 30 mm Hg) versus 1 of 126 patients with placebo ( < 1%).6


Rapid and sustained decreases in cardiac biomarkers of myocardial wall stress and injury were also observed in patients treated with mavacamten compared with placebo. At week 30, there was an 80% higher reduction in NT-proBNP in the mavacamten group than in the placebo group, whereas reduction in high-sensitive cardiac troponin I was 41% higher in the mavacamten group.6
Safety
The rate of treatment completion was 97%.6 Overall, 88% of patients in the mavacamten group and 79% in the placebo group experienced a treatment-emergent AE (TEAE), and most TEAEs were mild in severity.6 Three patients discontinued treatment because of AEs, 2 of whom were in the mavacamten group.6 Eleven serious AEs (SAEs) were reported in 10 patients in the mavacamten group compared with 20 SAEs reported in 11 patients in the placebo group. Serious cardiac AEs were reported in 4 patients receiving mavacamten (2 cases of AF and 2 of stress cardiomyopathy) and in 4 patients receiving placebo (3 cases of AF and 1 case of AF and congestive heart failure). One patient in the placebo group experienced sudden death.6
The change from baseline in systolic function (mean reduction in LVEF) observed in the mavacamten group was small, –3.9%, compared with –0.01% in the placebo group.6 Five patients (3 and 2 in the mavacamten and placebo groups, respectively) were subject to temporary discontinuation for LVEF less than 50% based on prespecified criteria in the study protocol; LVEF normalized in all 5 patients, who resumed treatment and completed the study. At week 30, an additional 4 patients in the mavacamten group experienced LVEF less than 50%, 3 of whom recovered after the 8-week washout period. The fourth patient underwent ablation for AF during the washout period and experienced a complication related to the procedure. This patient had a severe drop in LVEF followed by partial recovery to an LVEF of 50%.6
CONCLUSIONS
The results of the EXPLORER-HCM study showed that treatment with mavacamten met the efficacy and safety end points. In this first positive randomized phase 3 trial in patients with obstructive HCM, mavacamten showed improvement across comprehensive measures that assessed symptoms, function, health status, and levels of biomarkers of myocardial wall stress and injury that are correlated with long-term outcomes in HCM. Treatment with mavacamten was largely well tolerated, and mavacamten demonstrated a safety profile similar to that of placebo. The long-term efficacy and safety of mavacamten are currently being evaluated in the MAVA-LTE study. The results of EXPLORER-HCM support the benefits of mavacamten for targeted, disease-specific pharmacotherapy for obstructive HCM.6
REFERENCES
1. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-2779. doi:10.1093/eurheartj/ehu284
2. Ho CY, Olivotto I, Jacoby D, et al. Study design and rationale of EXPLORER-HCM: Evaluation of Mavacamten in Adults With Symptomatic Obstructive Hypertrophic Cardiomyopathy. Circ Heart Fail. 2020;13(6):e006853. doi:10.1161/CIRCHEARTFAILURE.120.006853
3. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379(7):655-668. doi:10.1056/NEJMra1710575
4. Cox S, O’Donoghue AC, McKenna WJ, Steptoe A. Health related quality of life and psychological wellbeing in patients with hypertrophic cardiomyopathy. Heart. 1997;78(2):182-187. doi:10.1136/hrt.78.2.182
5. Gersh BJ, Maron BJ, Bonow RO, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124(24):2761-2796. doi:10.1161/CIR.0b013e318223e230
6. Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10253):P759-P769. doi:10.1016/S0140-6736(20)31792-X
7. Mazzarotto F, Girolami F, Boschi B, et al. Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet Med. 2019;21(2):284-292. doi:10.1038/s41436-018-0046-0
8. Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138(14):1387-1398. doi:10.1161/CIRCULATIONAHA.117.033200


