
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
Addressing the Role of Edaravone in the Management of Amyotrophic Lateral Sclerosis and Gaps in Care and Access: Expert Panel Recommendations
Abstract
A virtual key opinion leader (KOL) and payer discussion was held on December 5, 2020. In attendance were 2 KOLs, both specialists in amyotrophic lateral sclerosis (ALS) at leading clinics in the United States, and 6 managed care executives from US regional health plans. The objective of this panel was to share opinions, ideas, and information around the treatment of ALS with edaravone, gaps in management and guidelines, and potential solutions. The panel concluded that coverage criteria for edaravone may need to be reassessed and treatment guidelines could be revisited to include a determination of place in therapy for edaravone.
Am J Manag Care. 2021;27(suppl 12):S231-S237. https://doi.org/10.37765/ajmc.2021.88732
Background
Amyotrophic lateral sclerosis (ALS) is a progressive, disabling, and fatal neurodegenerative disorder that causes muscle weakness, disability, and eventually death.1 It is most common in adults over the age of 60 years and affects 13,000 to 17,000 individuals in the United States with 5,000 new cases diagnosed yearly.2
Although the cause of most ALS is unknown, genetic mutations are the underlying cause of ALS in some patients. At this time, more than 40 genetic defects involved in various cellular pathways appear to cause the disease. This includes processing of ribonucleic acid molecules that may lead to ALS-related motor neuron degeneration, whereas other genetic mutations may cause defects in the natural process by which malfunctioning proteins are broken down and recycled to build new ones.3
Expansion in the C9orf72 gene is the most commonly known cause of both ALS and frontotemporal dementia with motor neuron-like inclusions. Clinical features of ALS and frontotemporal dementia may occur in both familial and sporadic cases and represent involvement of different brain regions from the same neurodegenerative process. The current hypothesis about gene and environmental interactions suggest that environmental factors can contribute, but no such factors have been established3; however, certain environmental factors are suspected to be potential risk factors. These include exposure to smoking, other toxins, or infectious agents; physical trauma; physical activity; certain diet and behavioral or occupational factors; and exposure to certain toxins.3
Diagnosis and Disease Assessment
ALS is primarily diagnosed based on symptom history, a physical exam, and a series of tests to rule out diseases with similar or overlapping symptoms.3 ALS is defined by the presence of both upper and lower motor neuron symptoms. Often, muscle and imaging tests such as electromyography and nerve conduction studies are used to exclude other causes of symptoms; these tests can show specific abnormalities that may suggest disorders such as peripheral neuropathy or myopathy rather than ALS.3


ALS Functioning Rating Scale Revised
In the last few decades, the ALS Functioning Rating Scale Revised (ALSFRS-R) has become a widely accepted measure of ALS progress, despite some limitations.4 This scale is a validated primary outcome measure used by clinical trialists and regulatory bodies to determine the rate of progression of disease. Point changes in ALSFRS-R can reflect clinically meaningful decline in functioning.4
The ALSFRS-R measures activity levels for 12 items, including speech, salivation, swallowing, handwriting, cutting food and handling utensils, dressing and hygiene, turning in bed, walking, climbing stairs, dyspnea, orthopnea, and respiratory insufficiency. A patient is evaluated by 5 levels per item (0,1,2,3,4) with 4 indicating the highest functionality and 0, the lowest functionality.
Japanese ALS Severity Classification Scale
Although ALSFRS-R is the widely used scale for measurement of functioning in patients with ALS, the Japanese ALS severity classification scale also defines function in patients with ALS and has been used as an inclusion criterion in clinical trials.5 This scale includes scores from 1 through 5, which are defined in Table 1.5
Patient Burden
Weakness is the defining symptom of ALS; it can affect essentially all volitionally controlled muscles. Early symptoms of ALS include muscle twitching and muscle cramps, loss of muscle control in hands and arms, impaired use of arms or legs, weakness and fatigue, tripping and falling, dropping items, slurred or thick speech, and difficulty projecting the voice.2 As the disease progresses, symptoms intensify and include shortness of breath, difficulty breathing, difficulty swallowing, and paralysis.2 ALS is typically relentless in progression, with a mean survival of 3 to 5 years from symptom onset.6
Economic Burden
The results of an analysis showed that the estimated national economic burden of ALS, including medical, nonmedical, and indirect costs, ranged from $256 million to $1.02 billion in 2010 US dollars.7 With an estimated increase of ALS cases, this burden is expected to increase, proportionally, 34% by 2040.7
In 2010, annual costs for ALS were estimated at $31,121 per patient. Current data from the Magellan Rx Management database show annual costs for patients with ALS, excluding those treated with edaravone, are similar, at $48,867 per patient in medical and pharmacy costs, excluding nonmedical and indirect costs associated with ALS. Updated total medical, nonmedical, and indirect costs are needed in order to accurately assess the value of novel therapies.
Current State of Treatment
No cure has yet been found for ALS. Currently, treatments aim to control symptoms, prevent complications, and improve quality of life.3 Care is best provided by a multidisciplinary team often comprising physicians; pharmacists; physical, occupational, and speech therapists; nutritionists; social workers; respiratory therapists; clinical psychologists; and home care and hospice nurses.3 A proper team can provide patients with individualized treatment plans and appropriate equipment to improve quality of life where possible.
Treatment plans for patients with ALS typically include a combination of drug therapy, physical therapy, speech therapy, nutritional support, and breathing support, depending on the patient’s disease progression and symptoms.3 At the time of this publication, 2 drug therapies are approved by the FDA for ALS: riluzole (Rilutek, Sanofi-Aventis) and edaravone (Radicava, Mitsubishi Tanabe Pharma America).
Treatment Guidelines
The American Academy of Neurology (AAN) released updated guidelines in 2009 and reaffirmed these guidelines in January 2020.8 According to the AAN, guidelines are affirmed when the methodology is still sound and either there is no new evidence or there is new evidence that would not change conclusions or recommendations. The guidelines include a level A recommendation for riluzole, meaning it should be offered to slow disease symptom progression in patients with ALS. When the guidelines were initially released, riluzole was the only FDA-approved drug for ALS, explaining why there was no mention of edaravone (it was approved in 2017); however, the guidelines were reaffirmed without modification after edaravone received FDA approval. The AAN does not currently have a publication discussing the role of edaravone in the treatment of ALS; practice parameter and quality measurement updates published since the most recent guidelines were released included no mention of edaravone.9,10 The lack of guidance around edaravone and its place in ALS therapy may raise questions around the AAN’s intent in omitting edaravone for payers when developing management strategies and outlining appropriate coverage criteria. Currently, there is an AAN ALS Quality Measures working group to revise a 2013 Neurology Quality Measures paper, which may address the role of edaravone in the treatment of ALS.11
The guidelines also include recommendations for nutritional therapies (enteral nutrition via percutaneous endoscopic gastrostomy should be considered to stabilize body weight and prolong survival) and respiratory therapies (noninvasive ventilation should be considered to treat respiratory insufficiency in ALS).8
The most recent guideline to address the place in therapy for edaravone and riluzole in patients with ALS was published in November 2020 in the Canadian Medical Association Journal. This guideline recommended the use of edaravone in a specific group of patients with ALS: those with a disease duration of less than 2 years, with a forced vital capacity (FVC) of 80%, a score of greater than 2 for all ALSFRS-R items, and a demonstrated steady decline in the ALSFRS-R over a 3 month interval.12
Overview of Treatment
Riluzole
Riluzole received FDA approval in 1995 for the treatment of ALS. The treatment is an oral tablet typically dosed at 50 mg twice daily. The first trial was a randomized, double-blind study of 155 patients with ALS; results showed riluzole had a significant effect on rate of survival and time to tracheostomy or death, which was about 90 days longer in the riluzole-treated group than in the control group.13
In a second trial, 959 patients were randomly assigned to 50 mg, 100 mg, or 200 mg of riluzole or placebo daily for 12 months in North America and for 18 months in Europe.13 The results showed no difference between the placebo group and the 50-mg group. However, improved early survival rates were observed in patients that received either 100 mg or 200 mg daily; the median survival time was 60 days longer in the riluzole group receiving 100 mg or 200 mg than in the control group.13 There was no difference observed in survival between the 100-mg and 200-mg riluzole groups; however, there was an increase in adverse events in the 200-mg group when compared with the 100-mg group. Adverse events associated with riluzole include asthenia, spasticity, nausea, and mild elevations in aminotransferase levels.13
Edaravone
The FDA approved edaravone in May 2017 for the treatment of ALS.14 Edaravone is an intravenous infusion administered by a health care provider or caregiver; the initial treatment cycle is 14 days of daily infusion, followed by a 14-day drug-free period.14 Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14-day drug-free periods. Adverse events associated with edaravone include bruising and gait disturbance, along with more serious risks such as hives, swelling, shortness of breath, and allergic reactions to sodium bisulfite.14
The initial phase 3 study, MCI186-16, failed to demonstrate efficacy of edaravone for ALS compared with placebo.15 A post hoc subgroup analysis was performed and focused on 2 newly defined subgroups: efficacy-expected subpopulation (EESP; patients with forced vital capacity [FVC] ≥80% and at least 2 points on all ALSFRS-R items) and greater-efficacy-expected subpopulation within EESP (dpEESP2y; onset of disease within 2 years).15 The post hoc analysis had a primary end point of change in ALSFRS-R score during the 24-week treatment period. The analysis showed efficacy in the dpEESP2y subgroup; change in baseline of the least-squares mean (± standard error values) in ALSFRS-R score was 2.20(±1.03) for EESP and 3.01(±1.03) in dpEESP2y.15
Based on the findings in the post hoc analysis of MCI186-16, the phase 3 MCI186-19 study had inclusion criteria of definite or probable ALS; normal respiratory function (defined as FVC≥80%); scores of at least 2 on all items of the ALSFRS-R; duration of disease of 2 years or less from symptom onset; aged 20 to 75 years; and deterioration of ALSFRS-R score during a 12-week pre-study observation period of 1 to 4 points.16 These inclusion criteria resulted in a homogeneous group of patients with a rapid rate of decline, where the functional rating scale was not expected to quickly have a floor effect. A total of 137 participants received either edaravone or placebo.
Results showed 33% less functional loss in the edaravone-treated group than in the placebo group in a 24-week period.16 Edaravone showed a clinically significant effect, with change from baseline in ALSFRS-R of 5.01(± 0.64) and −7.50(± 0.66) for edaravone and placebo, respectively.16 Over 6 cycles of treatment, 13% on edaravone vs 6% on placebo showed no decline in ALSFRS-R scores (0 points); 39% on edaravone vs 13% on placebo showed minimal function decline (<3points); and 9% on edaravone vs 24% on placebo showed significant functional decline (>9points).16
After the initial 24-week treatment period of MCI186-19, participants were given the option to enter an open-label extension of the study.16 A total of 123 patients opted to participate in the extension; 65 were previously given edaravone and 58 were previously in the placebo group. In a post hoc analysis of the extension study results, an estimate of likely progression in participants given the placebo vs edaravone in the initial treatment period was used to predict progression within each group with no change in treatment over the subsequent 24 weeks.16 Analysis of the projected progress compared with the actual rate of progression among those given edaravone for the full 48 weeks and those who switched from placebo to edaravone


showed that the projected decline for the placebo group was greater than the actual decline in the patients who switched from placebo to edaravone (13.0 vs 10.9 points, respectively).16 According to the FDA Center for Drug Evaluation and Research Clinical Review of edaravone, the results of the extension showed there was not a significant difference in increased disability using either the Japanese scale or the ALSFRS-R between the placebo- edaravone and edaravone-only patients.17
Gaps in Management
Current Payer Policies for Edaravone
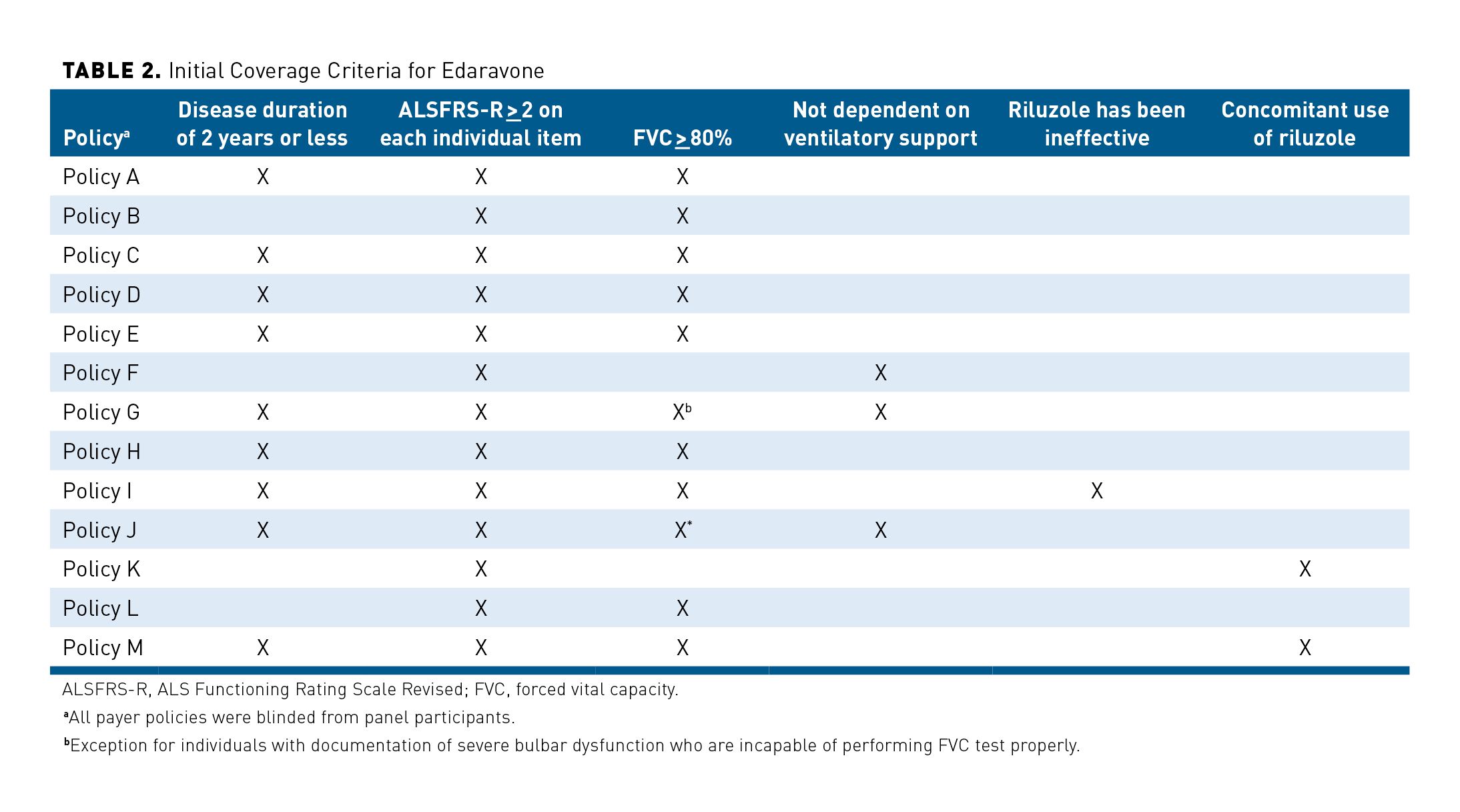
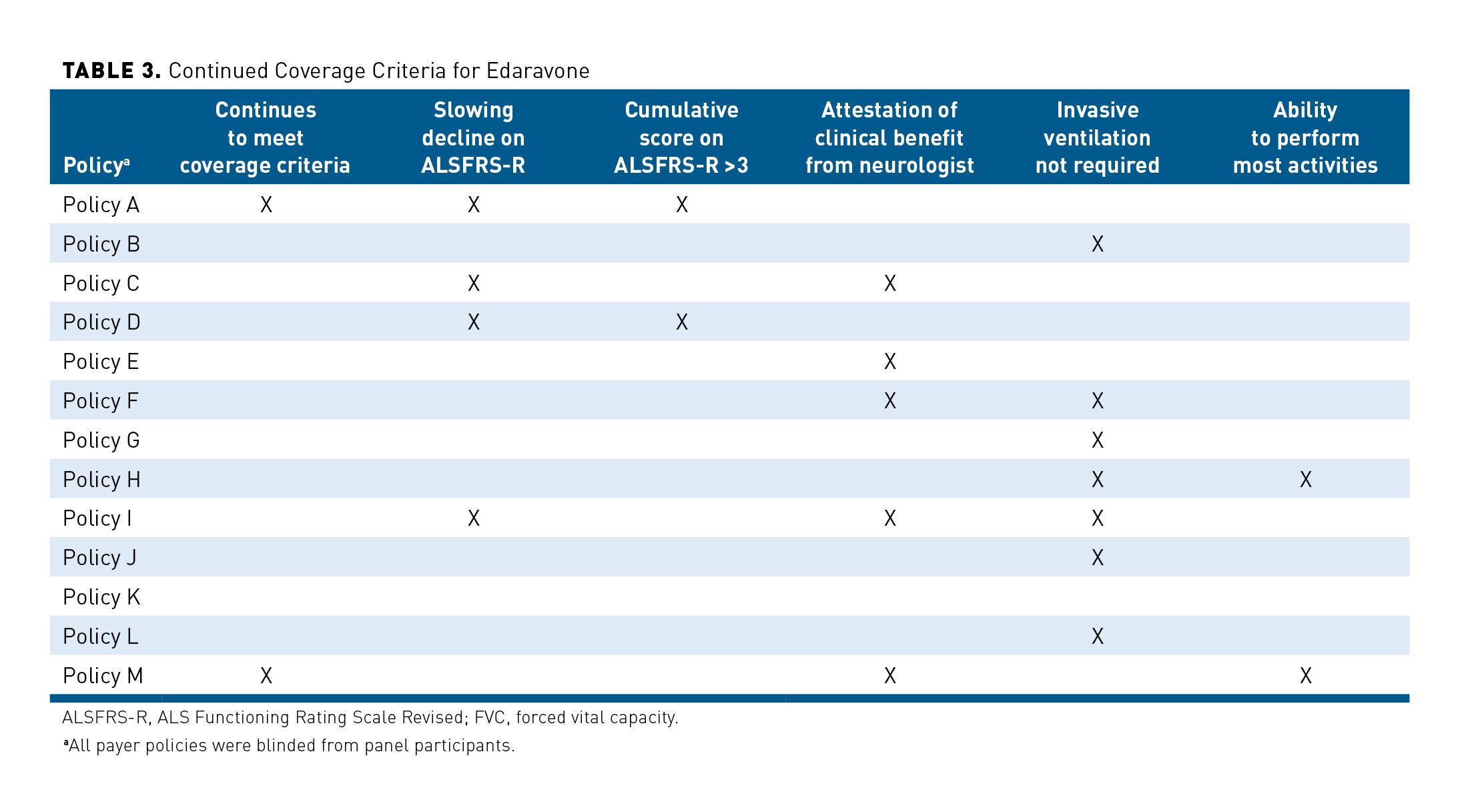
Edaravone is typically covered on the medical benefit and, across most policies, strict criteria must be met before coverage is provided, both initial and continued. The criteria generally align with the inclusion criteria for the phase 3 MCI186-19 study and include disease duration of 2 years or less, FVC of at least 80%, and an ALSFRS-R score of at least 2 on all items. However, the use of these criteria in addition to a wide variety of other criteria varies greatly among payers. The criteria for continued coverage differ as well; some of these criteria include neurologist attestation of clinical benefit, slowing decline of ALSFRS-R score (not defined), cumulative ALSFRS-R score of at least 3, and no requirement for invasive ventilation, or any combination of these. The lack of consensus across payer policy criteria for coverage and continuing coverage of edaravone may create a barrier for patients who could benefit from therapy. Table 2 and Table 3 show the disparity between criteria required for initial and continued coverage over a sampling of 13 payer policies.
Panel Insights and Recommendations
Policy Criteria
When discussing whether the coverage criteria warranted revisiting or modifying, the payer panelists noted that administrative burden is a main consideration. It is necessary to ensure that coverage criteria have real clinical purpose in defining an appropriate patient for therapy rather than merely creating a burden for all stakeholders—providers, payers, and patients. In assessing the 3 main coverage criteria, the discussion weighed the value of criteria inclusion against the administrative burden. Payers expressed that in this space, streamlining authorization and coverage processes and reducing burden will likely not result in additional patients being treated but could remove a barrier for providers, payers, and patients. Additionally, payers noted that in many cases these criteria are not absolute. If an individual patient’s provider feels the treatment will be beneficial, the case will be reviewed and the provider’s input will be taken into consideration.
Overall, although some payers did not feel changes would necessarily result in more patients being approved for treatment with edaravone, others agreed there is opportunity to revisit certain policy criteria for edaravone coverage to provide access for a broader patient population that could benefit from therapy.
Disease Duration
The majority of the panel agreed that the requirement of disease duration of 2 years or less warranted revisiting. The KOLs expressed that limiting access to treatment based on disease duration did not have clinical value. Specifically, because of the variation in

disease progression, a patient diagnosed young with slow disease progression could meet all coverage criteria except for disease duration but could be prohibited from coverage. The KOLs also noted that disease onset cannot be accurately assessed in many patients with ALS.
Based on evidence in the post hoc analysis of MCI186-19, several payers were convinced that the population of patients who may benefit from edaravone extends beyond those with a disease duration of 2 years or less. Data from the MCI186-19 trial suggests that the functional decline in patients who switched to edaravone after receiving the placebo was comparable to those who were initiated on edaravone earlier in disease progression.17 There was overall consensus that an opportunity exists to reassess this criterion and to determine when a patient should start therapy to benefit from it clinically. At this point, most payers agreed the current disease duration criterion creates a gap for patients who could still benefit from treatment by flattening the scope of linear disease progression.
FVC of At Least 80%
There was consensus among a majority of the panel that removal of the FVC criterion from coverage policies should be considered. One of the payer participants noted that their respective health plan had already removed the criterion based on available literature. Results from a post hoc analysis of MCI186-19 demonstrated that ALS patients with FVC of 80% received significant benefit from treatment with edaravone, indicating that baseline FVC may not be an appropriate criteria for determining patients eligible for treatment.18 Overall, the KOLs and most payers agreed that inclusion of this criterion did not effectively define a patient population and may not be a necessary threshold to determine coverage.
Even where a small minority did not agree that the FVC criterion should be removed from coverage policies, they did agree that an exception for patients with severe bulbar dysfunction may be appropriate. A bulbar dysfunction exception is currently present in some policies having a requirement of FVC of at least 80%. This exemption can apply to individuals with documentation of severe bulbar dysfunction who are incapable of properly performing an FVC test.
ALSFRS-R Score of At Least 2 on All Items
The KOLs expressed that requiring a specific score on each individual item of the ALSFRS-R is an outcome measure–related issue and not applicable in clinical care. The reason this inclusion criterion was included in clinical trials was to prevent complete loss of patients on ALSFRS-R items (ie, floor effect); results from a post hoc analysis of MCI186-16 showed efficacy among early stage ALS patients, or those with scores of at least 2 points on ALSFRS-R items.19 Thus, the inclusion criteria for MCI186-19 was determined accordingly.19 Analysis of MCI186-19 results suggest that edaravone has a clinically meaningful effect in retaining function for ALS patients.20 Specifically, there was a notable reduction in patients experiencing a decline in ALSFRS-R score from 3 to 2 for items including swallowing, eating motion, walking, and climbing stairs.20 These findings suggest edaravone can play a role in preserving function in some patients with ALS, but this would be restricted if access is limited to those with only specific ALSFRS-R scores. Applying a score across all items can create a challenge for patients with moderate disease progression who have a delayed diagnosis. KOLs explained that efficacy is observed in patients who receive edaravone with progressed disease; patients who may not meet the required score on all ALSFRS-R items may still receive a clinical benefit from treatment.
Across the payer participants, there was agreement that a measure needs to be in place to determine the patients who would most benefit from treatment. Currently, payers feel the ALSFRS-R is the most evidence-based measure; however, several payers expressed interest in revisiting these criteria to increase flexibility. One option discussed was weighting different items of the ALSFRS-R score more than others, perhaps based on which items are more closely linked to clinical benefit with edaravone.
Payers also expressed that ALSFRS-R items could indicate the ability of patients to live independently; having this requirement in place would allow clinical benefit, or slowing decline, to be better quantified. Data suggests that early intervention with edaravone is associated with a reduction of occurrence of death, tracheostomy, permanent assisted ventilation, and hospitalization in patients with ALS.21 However, KOLs noted that in terms of determining a patient’s ability to live independently, the Japanese ALS severity classification could be a more accurate measurement. Requiring a patient to have a specific score on this very simple functional scale could better classify which patients are able to live independently.
Continued Coverage Criteria for Edaravone
As previously noted, continued coverage policies vary more widely than initial coverage policies. In some cases, the KOLs noted there does not seem to be clinical evidence to justify some of the criteria that certain policies include. Certain payers were in line with KOLs and viewed some continued coverage criteria as irrelevant. These payers felt that reauthorization policies could be streamlined to reduce administrative burden. Some payers noted they would keep 2 criteria: (1) invasive ventilation is not required, and (2) the ability to perform most daily activities. However, most payers noted that, aside from cases where invasive ventilation is required, their review process takes the individual’s progress while being treated with edaravone into account to make an informed decision on coverage continuation.
There was consensus among the group that the most important criterion for continued coverage is an attestation of clinical benefit from the treating neurologist. Certain payers felt that, along with a neurologist attestation, some documentation of slowing decline should be included to justify continued treatment. This documentation and attestation will be considered in making a coverage decision and, based on the individual patient needs, may carry more weight toward approval than policy renewal criteria alone.
Payers did note it would be beneficial to have a clearer determination of which measures define when a patient will continue to benefit from treatment with edaravone. KOLs noted that if there is still motor function to be preserved, most patients will benefit from continuation but in practice, they also consider the patient’s burden of treatment with an infused medication. Evidence does suggest edaravone is associated with reduction in decline in ALSFRS-R scores.20 Payers agreed they would look for an attestation from the neurologist when reviewing a patient’s request to continue therapy.
Guidelines for Treatment of Patients With ALS
As the treatment landscape for ALS changes, the KOLs noted there is an ongoing effort by specialists in the ALS space to update guidelines, and there is a current need for a consensus guideline to be reviewed and endorsed by the AAN and the American Association of Neuromuscular and Electrodiagnostic Medicine (AANEM). An endorsement of consensus could be looked at in lieu of updates made during a review of the current reaffirmed guidelines from 2019.
Payers discussed that while, in their experience, guidelines are not always referenced in policies, the guidelines are 1 resource used to help determine appropriate coverage criteria. Some payers noted that the guidelines are second to FDA approval criteria in terms of evidence to help structure policies and, in the absence of complete comprehensive guidelines in a treatment space, designing an appropriate, evidence-based policy is challenging. A consensus guideline would be valuable in reducing treatment variation and barriers to treatment and in increasing consistency in coverage and management.
Although policies can be designed and structured based on available literature, data, and evidence, a resource such as a guideline or an expert consensus statement would be helpful and substantive in determining a clearly defined patient population appropriate for therapy. Payers noted that guidelines are not a necessity in developing policies, but when available, payers will refer to and rely on them as a significant resource.
Cost Effectiveness in ALS Treatment
Overall, payers noted that cost-effectiveness measures and data would not weigh heavily in coverage decisions for this therapy because the total budget impact is not significant. KOLs discussed that many patients choose not to start edaravone therapy or choose to discontinue therapy because the benefit of slowed disease progression does not outweigh the burden of treatment administration via infusion. As the population of patients with ALS is already a small fraction of the payers’ member populations, the budget impact of the small number of patients receiving edaravone would be insignificant enough that cost and cost-effectiveness evaluations would not weigh in the decision-making process in a meaningful way.
Future Needs and Considerations
Moving forward, the participants concluded there is a need to approach the AAN and AANEM for a reassessment of the guidelines, ensuring that a clear stance on all available treatments is included. These guidelines will create a sense of understanding and may allow for cohesiveness across policies, helping to reduce barriers to treatment and unnecessary burden on stakeholders.
At the time of the panel, payers noted that real-world outcome data would be ideal in helping to determine broader patient populations that could benefit from treatment, outside of those who meet the coverage criteria. Data could support a clear definition of patients for initial treatment as well as those for whom continued treatment would be appropriate and clinically valuable. In May 2021, real-world evidence presented at the European Network for the Cure of ALS demonstrated that continued edaravone treatment may improve overall survival in patients with ALS when compared with ALS patients treated predominantly with riluzole.22 Additional, similar real-world outcomes could be used to support clear guidelines and determinations on the role of edaravone in ALS therapy.
The panel concluded that the available evidence coupled with the diverse starting and continuation policy criteria warranted a reassessment of coverage criteria with the possibility of certain criteria being removed, such as the disease duration and FVC criteria, and other criteria being restructured, such as ALSFRS-R scores, and that treatment guidelines from organizations like the AAN would be useful and valuable in helping to better structure coverage policies to ensure access for appropriate patients. Overall, clear guidance and consensus on the role of edaravone in the treatment of ALS should lead to more consistent policy criteria for starting and continuation of therapy, leading to appropriate access to care for patients with ALS.
Acknowledgements
The authors acknowledge Haita Makanji, PharmD, and Stacy Inman, PharmD, for facilitating the panel discussion and providing writing assistance; and Carole Kallas for providing administrative support.
Author affiliations: Priority Health, Grand Rapids, MI (CJB); Independent Health, Buffalo, NY (MB); Magellan Health, Scottsdale, AZ (CC); Georgetown University Medical, Washington, DC (JC); CenCal Health, Santa Barbara, CA (JJ); BlueCross BlueShield of Western New York, Buffalo, NY (LL); Mayo Clinic, Jacksonville, FL (BO); Moda Health, Portland, OR (CR); Magellan Rx Management, Middletown, RI (LS); Health New England, Springfield, MA (GT).
Funding source: Financial support for this work was provided by Mitsubishi Tanabe Pharmaceuticals and facilitated by Magellan Rx Management.
Author disclosures: Dr Barrington, Burruano, Dr Choi, Dr Januska, Dr Lachuk, Dr Oskarsson, Dr Rodriguez, Speicher, and Dr Tereso have received honoraria from Magellan Rx Management; Dr Choi has also received grants from Massachusetts General Hospital, Washington University, and Biogen; Dr Oskarsson has held board membership at The ALS Association, Western ALS Study Group, and ALS Home Health Medical Standards Group; Dr Oskarsson has also received grants from Mitsubishi Tanabe Pharma, MediciNova, Inc, Biogen, Cytokinetics, AZ Therapeutics, Eisai, Roche, and GlaxoSmithKline; Dr Oskarsson has also received honoraria from Mitsubishi Tanabe, MediciNova, Inc, Biogen, and Biohaven, Tsumura & Co.; Dr Carney and Speicher have been employed at Magellan Rx Management.
Authorship information: Concept and design (CJB, MB, CC, JC, JJ, LL, CR, LS, GT); Acquisition of data (CC, JJ, LL, BO, LS); Analysis and interpretation of data (CJB, MB, CC, JC, LL, BO, CR, LS, GT); Drafting of the manuscript (CJB, MB, JC, LL, BO, CR, LS, GT); Critical revision of the manuscript for important intellectual content (JJ, LS); Administrative, technical, or logistic support (LS).
Address correspondence to: Lindsay Speicher, JD. Email: lspeicher@ magellanhealth.com
REFERENCES
1. Maragakis NJ, Galvez-Jimenez N. Epidemiology and pathogenesis of amyotrophic lateral sclerosis. UpToDate. Updated September 26, 2011. Accessed May 20, 2021. https://somepomed.org/articulos/ contents/mobipreview.htm?25/30/26081?source=see_link&anchor=H5
2. Johns Hopkins Medicine. ALS – amyotrophic lateral sclerosis. Accessed May 20, 2021. https://www. hopkinsmedicine.org/neurology_neurosurgery/centers_clinics/als/conditions/als_amyotrophic_lateral_ sclerosis.html
3. National Institute of Neurological Disorders and Stroke. Amyotrophic Lateral Sclerosis (ALS) fact sheet. National Institutes of Health. Updated June 20, 2020. Accessed May 20, 2021. https://www.ninds.nih.gov/ Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet
4. Nicholson KA, Cudkowicz ME, Berry JD. Clinical trial designs in amyotrophic lateral sclerosis: Does one design fit all? Neurotherapeutics. 2015;12(2):376-83. doi:10.1007/s13311-015-0341-2.
5. Sato Y, Nakatani E, Watanabe Y, et al. Prediction of prognosis of ALS: Importance of active denervation findings of the cervical-upper limb area and trunk area. Intractable Rare Dis Res. 2015;4(4):181-189. doi:10.5582/irdr.2015.01043
6. Chio A, Logroschino G, Hardiman O, et al. Prognostic factors in ALS: A clinical review. Amyotroph Lateral Scler. 2009;10(5-6):310-323. doi:10.3109/17482960802566824
7. Santaniello B. ALS managed care considerations. Am J Manag Care. 2018;24(suppl 15):S336-S341.
8. American Academy of Neurology. AAN summary of evidence-based guideline for clinicians: The care of the patient with amyotrophic lateral sclerosis: drug, nutritional and respiratory therapies. American Academy of Neurology. Updated January 11, 2020. Accessed May 20, 2021. https://www.aan.com/ Guidelines/home/GuidelineDetail/370
9. Miller RG, Brooks BR, Swain-Eng RJ, et al. Quality improvement in neurology: Amyotrophic lateral sclerosis quality measures: Report of the quality measurement and reporting subcommittee of the American Academy of Neurology. Neurology. 2013;81(24):2136-40. doi:10.1212/01.wnl.0000437305.37850.f9
10. Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227-33. doi:10.1212/WNL.0b013e3181bc01a4
11. American Academy of Neurology. Quality Measures Neuromuscular. Accessed June 17, 2021. https:// www.aan.com/policy-and-guidelines/quality/quality-measures2/quality-measures/neuromuscular/
12. Shoesmith C, Abrahao A, Benstead T, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. CMAJ. 2020;192(46):E1453-E1468. doi:https://doi.org/ 10.1503/cmaj.191721
13. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;2012(3):CD001447. doi:10.1002/14651858.CD001447.pub3.
14. FDA approves drug to treat ALS. FDA. Updated March 28, 2018. Accessed May 20, 2021. https://www. fda.gov/news-events/press-announcements/fda-approves-drug-treat-als
15. EDARAVONE (MCI-186) ALS 16 STUDY GROUP. A post-hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(suppl 1):11-19. doi:10.1080/21678421.2017.1363780
16. Shefner J, Heiman-Patterson T, Pioro EP, et al. Long-term edaravone efficacy in amyotrophic lateral sclerosis: post-hoc analyses of Study 19 (MCI186-19). Muscle Nerve. 2020;61(2):218-221. doi:10.1002/ mus.26740
17. Breder, CA. FDA Center for Drug Evaluation and Research (CDER) Clinical Review 209176Orig1s000. Accessed June 8, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/ nda/2017/209176orig1s000medr.pdf
18. Pattee G, Suarez Zambrano G, Zhang J, et al. Post hoc analysis of edaravone study 19: efficacy in bulbar onset ALS patients with and without reduced pulmonary function. Presented at: Muscular Dystrophy Association MDA Virtual Clinical & Scientific Conference; March 21-25, 2020; virtual. Poster 53. Accessed June 8, 2021. https://www.mdaconference.org/node/942.
19. Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505-512. doi:10.1016/S1474-4422(17)30115-1.
20. Palumbo J, Apple S, Agnese W, et al. Retaining physical function in amyotrophic lateral sclerosis with edaravone: Post hoc analysis of pivotal study MCI186-19. CLT-39 The NEALS Consortium - a collaborative research organization. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(suppl 1):262-288. doi:10.1080/21678421.2019.1646997
21. Brooks B, Sakata T, Takahashi T, et al. Early intervention with edaravone in study 19 was associated with decreased hospitalization, tracheostomy, ventilation, and death in patients with ALS. Presented at: Muscular Dystrophy Association MDA Virtual Clinical & Scientific Conference; March 21-25, 2020; virtual. Poster 57. Accessed June 8, 2021. https://mdaconference.org/node/1148
22. Brooks BR, Ciepielewska M, Zhang J, et al. Continued intravenous edaravone treatment of ALS patients increases overall survival compared with no IV edaravone treatment in a US administrative claims database. Presented at: ENCALS meeting; May 12-14, 2021; virtual. https://www.encals.eu/wpcontent/uploads/2019/05/ENCALS-2021_ABSTRACT-BOOK.pdf.


