
- Center on Health Equity & Access
- Clinical
- Health Care Cost
- Health Care Delivery
- Insurance
- Policy
- Technology
- Value-Based Care
A Decade of Progress: Advances in the Third-Line Treatment of Patients With Metastatic Colorectal Cancer
Abstract
The treatment of metastatic colorectal cancer (mCRC) remains challenging. There has been substantial progress in understanding the molecular pathology of the disease that has led to meaningful advancements in treatment options with varying mechanisms of action, although treatment remains costly. Cytotoxic therapies, which are typically combined with targeted therapies, remain the mainstay of first- and second-line treatment for mCRC. While also relevant in earlier lines of therapy, molecular testing has become increasingly important to guide therapy across lines of therapy, for which treatment options are limited. A paucity of data exists in establishing clinical criteria for optimizing the sequencing of therapies in the third line and beyond. A customized approach should consider the efficacy of the therapy balanced with the patient’s goals. Sequencing criteria should include a consideration for exposing patients to as many different modes of therapy as possible while preserving quality of life, avoiding serious toxicities, and accounting for the potential impact of cumulative toxicities from prior therapies.
Am J Manag Care. 2024;30:S23-S30. https://doi.org/10.37765/ajmc.2024.89545
For author information and disclosures, see end of text.
Introduction
Colorectal cancer (CRC) is the third most common cancer type in the United States. It accounts for 8% of all new cancer cases for both males and females, with an estimated 153,000 new cases diagnosed in 2023.1 That year, CRC was the third leading cause of cancer death when stratified by sex, with over 52,000 expected CRC-related deaths reported in men and women combined.1
The rate of CRC diagnosis remains high; however, both incidence and mortality rates have declined over past decades. For example, the incidence rate was 38.7 per 100,000 people in 2016 as compared with 60.5 per 100,000 people in 1976; this reduction was accompanied by a decrease by more than 50% in peak mortality rates over those 40 years.2 These improvements largely can be attributed to improved screening, better treatments, and the identification of biomarkers.2 Despite this success, there is a discrepancy among age groups, in which patients 65 years or older have a decreasing incidence rate of 3.3% annually, but patients younger than 50 years have an increasing incidence rate of 2% annually (and a 1.3% annual increase in death rates).2
At the time of CRC diagnosis, as many as 23% of patients have metastatic CRC (mCRC).3 An estimated 25% to 50% of patients diagnosed with lymph node–positive CRC also may develop metastases eventually.4 Outcomes for patients with CRC become progressively worse when cancer becomes metastatic, with a 5-year survival rate of 15.6% for patients with mCRC compared with 91% for those with localized disease.3
Economic Impact of mCRC
CRC is the malignancy associated with the second highest costs, accounting for $24.3 billion in national health care expenditures in 2020 per the National Institutes of Health.5 According to Medicare Parts A and B claims for billed services that year, $23.7 billion was spent on medical services (eg, hospitalizations, outpatient hospital services, infusion and injectable drugs, hospice care, and home health care).5 In addition, as estimated using Medicare Part D claims, $600 million was spent on oral prescription drugs.5
Few recent studies have been conducted to evaluate the costs related to mCRC and its management in the United States.
One systematic review of direct costs related to treating mCRC was conducted in 2022.6 While limited by the inclusion of international studies, the review included US-based studies (14 of 29) and converted all costs to 2018 US$. The estimated cost of treatment for a patient with mCRC broadly ranged from $12,346 to $293,461; the wide range was attributable largely to the heterogeneity of treatment. The same study found that estimated average costs for patients receiving systemic therapy could be as high as $300,000 due to the costs of biologic agents. A major cost driver in the treatment of patients with metastatic disease is the treatment of hematologic adverse events (AEs), such as neutropenia, thrombocytopenia, and/or anemia, associated with cytotoxic treatment.7
Review of Molecular Pathology
Roughly 85% of CRCs arise from the alteration in the number and structure of chromosomes known as chromosomal instability. The most clinically relevant pathways for chromosomal instability colorectal tumors are through the Wnt and MAPK pathways. When CRC develops through the Wnt pathway, Wnt proteins bind to a Frizzled family receptor, leading to β-catenin accumulation and hence transcription activation and uncontrolled cell proliferation. In MAPK-activated CRC, receptor tyrosine kinases are activated. Signaling molecules such as RAS and RAF thus become constitutively active, resulting in uncontrolled cell proliferation.8
The remaining 15% of CRCs that are not attributed to chromosomal instability arise as a result of microsatellite instability (MSI), a general instability of DNA sequences (known as microsatellites) resulting from either mutation of a mismatch repair (MMR) gene or hypermethylation and subsequent silencing of the MLH1 promoter.8 In the absence of any of the 5 MMR gene mutations of interest (ie, mutation of MLH1, MSH2, MSH6, or PMS2 or hypermethylation of MLH1), the tumor is considered microsatellite stable (MSS). If at least 2 of these MMR genes are mutated, the tumor is considered MSI-high (MSI-H).8 This distinction between MSS and MSI-H disease is crucial—it substantially impacts the treatment choice for mCRC.
Molecular Testing
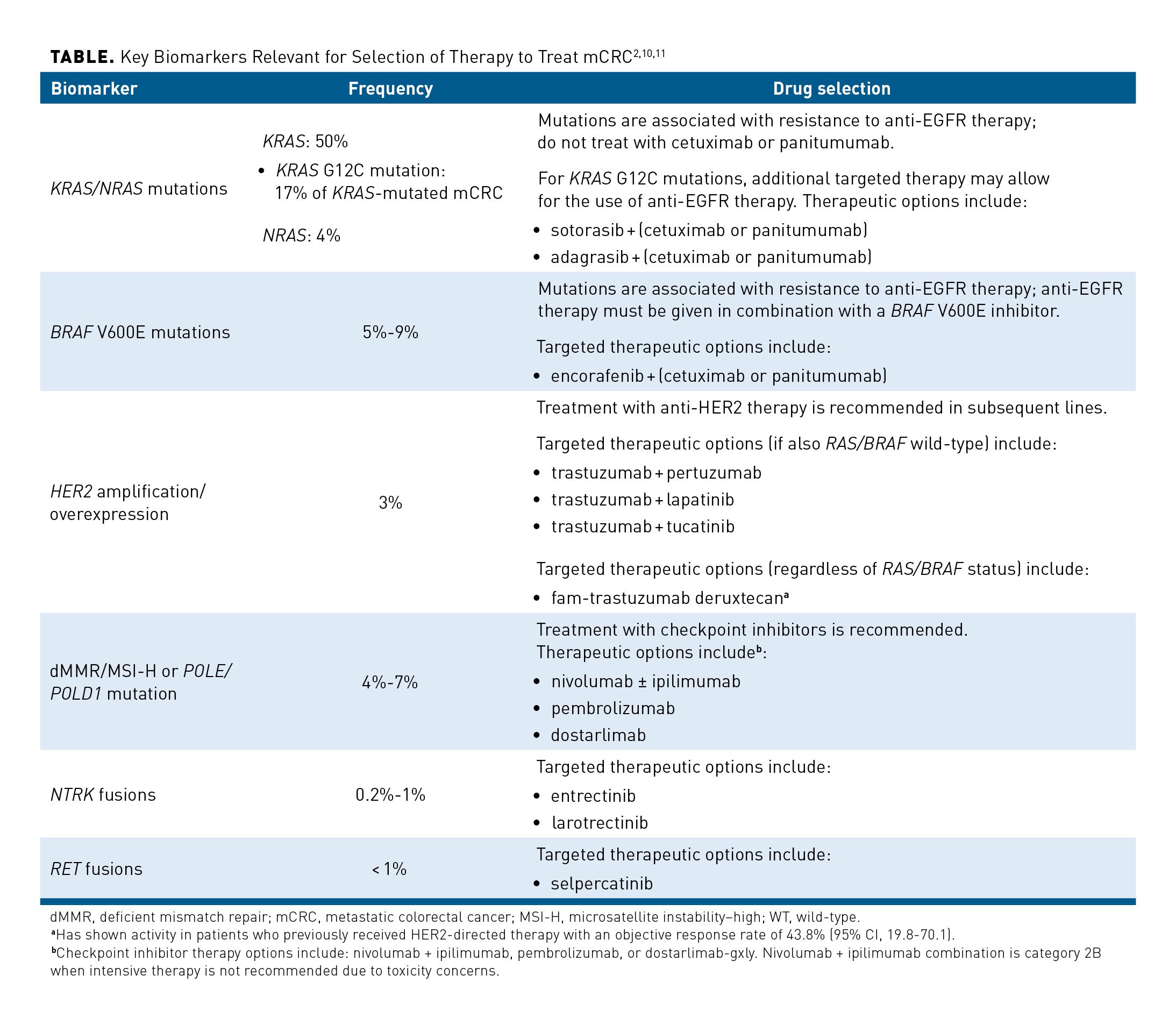
Molecular testing has accelerated the development of biomarker-targeted therapies for CRC, and it may improve outcomes of patients with metastatic disease.9 If biomarker testing indicates the presence of identifiable, targetable defects, then therapy can be tailored for that patient.2 The Table summarizes the range of biomarkers that may be relevant in guiding therapy for patients with mCRC.2,10,11 The 2024 NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Colon Cancer (version 1.2024) include checkpoint inhibitor therapy options: nivolumab ± ipilimumab, pembrolizumab, or dostarlimab-gxly. Nivolumab + ipilimumab combination is category 2B when intensive therapy is not recommended due to toxicity concerns.2
Activation of EGFR, a family of receptors including HER2, can trigger a series of downstream signaling pathways leading to cellular proliferation; EGFR is overexpressed in as many as 77% of patients with CRC.9 The oncogenes KRAS and BRAF are major contributors to the chromosome instability found in 85% of CRC cases and are associated with resistance to anti-EGFR therapy.2,9
For patients with mCRC, the 2024 NCCN Guidelines in Oncology Colon Cancer (version 1.2024) and Rectal Cancer (version 1.2024) currently recommend broad molecular profiling; this should include testing for common biomarkers (eg, mutations in KRAS/NRAS and BRAF, amplification of HER2, and MSI) and rare biomarkers (eg, mutations in POLE/POLD1, RET, and NTRK).2,12


Current Treatment Landscape for mCRC in the First and Second Line
Current treatment pathways for patients with unresectable mCRC involve multiple lines of therapy—and these pathways have changed dramatically over the past few decades with the advent of biologic agents and immunotherapy. First-line (1L) treatment typically involves a combination of 1 or more chemotherapeutic agents (eg, FOLFOX [leucovorin, fluorouracil, and oxaliplatin], or FOLFIRI [leucovorin, fluorouracil, and irinotecan]) with or without a monoclonal antibody.2 In the absence of targetable mutations, this is followed by second-line (2L) treatment with a different chemotherapeutic regimen (typically, the alternative 1L option that was not given initially) with or without a monoclonal antibody.2
Anti-VEGF Therapy
VEGF (in particular, VEGF-A) expression is increased in CRC, and this increase plays an important role in promoting tumor angiogenesis and, subsequently, tumor growth and metastases.13 Bevacizumab, a VEGF-A inhibitor, was approved in 2004 for the treatment of mCRC; it prevents the interaction of VEGF with its receptors, reduces microvascular growth, and inhibits metastatic disease progression.14 The addition of bevacizumab to chemotherapy has shown benefit in both progression-free survival (PFS) and overall survival (OS) across multiple lines of therapy.15-17
Anti-EGFR Therapy
Treatments for mCRC can involve targeting the EGFR pathway through use of anti-EGFR monoclonal antibodies. Cetuximab, approved by the FDA in 2004, binds to the external domain of EGFR and promotes internalization and degradation of the receptor.9,18 Panitumumab, another EGFR inhibitor indicated for treatment of mCRC, was approved in 2006.19 By binding specifically to EGFR and competitively inhibiting the binding of other ligands, panitumumab prevents ligand-induced receptor autophosphorylation and activation of receptor-associated kinases. However, anti-EGFR therapies are not recommended for patients with tumors with RAS or BRAF mutations downstream signaling pathways, which causes resistance to them.9,20 Additionally, the presence of HER2 amplification or right-sided tumor location appear to predict for resistance to anti-EGFR therapies.21,22
Immunotherapy for dMMR/MSI-H or POLE/POLD1 mutation
In 2020, pembrolizumab, a PD-1 targeting humanized immunoglobulin G4 (IgG4) antibody, was FDA-approved for the treatment of patients with MMR deficiency (dMMR)/MSI-H CRC. Pembrolizumab binds to PD-1 and prevents its interaction with PD-L1 and PD-L2. By doing so, it releases PD-1 pathway-mediated inhibition of immune response, including the antitumor immune response.23 Additional NCCN Guidelines preferred PD-1 blockers for dMMR/MSI-H mCRC include nivolumab and dostarlimab.2 Additionally, nivolumab can also be used in combination with ipilimumab in either the 1L or later lines of therapy as recommended by the NCCN Guidelines.2,24
Third-Line Treatment Options
Improvements in the efficacy of therapies and the number of lines of therapies available are among possible factors that have improved treatment outcomes for mCRC.25 A growing number of patients with mCRC who receive 1L and 2L chemotherapy are treated with third-line (3L) or subsequent lines of therapy (3L+). This trend highlights the importance of optimizing the selection and sequencing of later-line treatments for these patients.26
Best practices for treating patients with mCRC in the 3L are typically made based on considerations of clinical guidelines, patient-specific characteristics (eg, performance status, medical history), disease-specific characteristics (eg, molecular biomarkers, previous therapies received), effective AE management, and patient quality of life (QOL).26-28
Given the complexity and patient-specific nature of these treatment decisions, clinical pathways should allow for flexibility for any FDA-approved therapy in the 3L setting and beyond.
Patients who experience relapse of disease after prior lines of treatment have a limited number of alternatives. One is treatment with cetuximab or panitumumab for any patient who has not previously received these drugs; this, however, is a less common 3L treatment, as these are typically administered in an earlier line of therapy. If the mCRC is dMMR/MSI-H or harbors the POLE/POLD1 mutation, immunotherapy can still be considered if it was not received in a previous line; however, similar to anti-EGFR therapies, immunotherapies are often used earlier in a patient’s treatment course.2 Another option is to leverage biomarker-targeting therapies (eg, agents targeting HER2, BRAF V600E, NTRK, and RET [Table]).2,27
In addition, patients with mCRC in the 3L have access to several therapies in different classes that are indicated specifically for the 3L and beyond. These include yet another chemotherapeutic agent (ie, trifluridine/tipiracil [FTD/TPI], given alone or with bevacizumab) or an agent other than a chemotherapeutic drug (eg, regorafenib, fruquintinib).2 Recognition that repeated use of chemotherapies can be associated with substantial AEs, such as myelosuppression, has fueled interest in treatment regimens used in the 3L that offer improved safety and tolerability.29
Regorafenib
Regorafenib, an oral multikinase inhibitor approved by the FDA in 2012, remains a core standard of care option for patients who have been previously treated for mCRC. In addition to its other indications in hepatocellular carcinoma and metastatic gastrointestinal stromal tumor, regorafenib is indicated to treat patients with mCRC who have been previously treated with chemotherapy (including a fluoropyrimidine, oxaliplatin, and irinotecan) and biologic therapies (ie, an EGFR inhibitor [if RAS wild-type] or a VEGF inhibitor).30 Regorafenib has a unique mechanism of action; it inhibits at least 20 membrane-bound and intracellular kinases involved in oncogenesis, tumor angiogenesis, metastasis, and tumor immunity. Kinases involved are RET, RAF, BRAF, and VEGFR, among others.30
The efficacy of regorafenib was demonstrated in the multicenter, randomized, double-blind, placebo-controlled CORRECT trial (NCT01103323) that evaluated OS, PFS, objective tumor response rate, and disease control rate in 760 patients with previously treated mCRC.31 In this trial, patients were randomly assigned 2:1 to receive 160 mg once daily of regorafenib plus best supportive care (BSC) (505 patients) or placebo plus BSC (255 patients) on days 1 through 21 of a 28-day cycle.
Patients receiving regorafenib plus BSC demonstrated statistically significant improvements in both OS and PFS when compared with patients receiving placebo plus BSC. Patients experienced OS that was 1.4 months longer than noted in those given placebo (HR, 0.77; 95% CI, 0.64-0.94; P = .0052) and PFS that was 0.2 months longer than seen in those given placebo (HR, 0.49; 95% CI, 0.42-0.58; P < .0001).31 Results of a phase 3 trial of 204 patients at study sites throughout China, Hong Kong, South Korea, Taiwan, and Vietnam showed similar results.32
The rate of AEs in the CORRECT trial was high, with 93% of patients experiencing an AE of any grade in the regorafenib arm.31 The most common any-grade AEs included fatigue (47%), hand-foot skin reaction (HFSR) (47%), diarrhea (34%), and anorexia (30%). HFSR and fatigue of at least grade 3 were observed in 17% and 10% of patients, respectively. Notably, incidence of myelosuppression of grade 3 or more was low with regorafenib (grade 4, ≤ 5%; grade 4, ≤ 1%).30 Dose reduction or interruption was needed for 67% of patients receiving regorafenib vs 23% of those given placebo.31 AEs resulting in discontinuation occurred in 8.2% of patients receiving regorafenib compared with 1.2% of those receiving placebo, with HFSR and rash being the most common reasons for permanent discontinuation of regorafenib.30
Based on the rate of AEs in the CORRECT trial, the multicenter, phase 2, open-label ReDOS trial (NCT02368886) was initiated in 39 outpatient cancer centers in the United States to explore dose optimization strategies for regorafenib.33 Enrollment in this study occurred between June 2, 2015, and June 22, 2017; patients were randomly assigned to receive 80 mg once daily of regorafenib orally with weekly dose-escalation or a steady dose of 160 mg once daily. In both groups, regorafenib was administered for 21 consecutive days of a 28-day cycle. During subsequent cycles, patients received the highest tolerated dose from cycle 1, up to 160 mg. The primary end point of this study was the percentage of patients able to complete 2 full cycles of treatment and begin the third cycle. Secondary end points included efficacy end points such as PFS, OS, time to progression, as well as cumulative dose received within the first 2 cycles, proportion of patients with HFSR of at least grade 3, and QOL measures.
The dose-escalation strategy resulted in 43% of patients (95% CI, 29%-56%) successfully initiating treatment cycle 3 as compared with 26% of patients (95% CI, 15%-37%) starting with full-dose therapy; the difference was significant (P = .043).33 The main factor preventing patients from starting a third cycle was progression of disease (dose-escalation group, 37%; standard-dose group, 47%).
There was a 3.8-month difference in OS between the dose-escalation group and the standard-dose group (9.8 vs 6.0 months; HR, 0.72; 95% CI, 0.47-1.10; log-rank P = .12). Additionally, the incidence of grade 3 AEs commonly associated with regorafenib treatment—specifically fatigue, HFSR, and hypertension—were numerically lower in the dose-escalation group than in the standard-dose group. This numerical reduction in the incidence of fatigue, HFSR, and hypertension was further supported by the results of another phase 2 study that evaluated a slightly modified dose-escalation strategy that started at 120 mg daily.34 Overall, these data suggest that more patients are able to tolerate regorafenib with a dose-escalation strategy of regorafenib.
Based on results from these trials, the NCCN Guidelines recommend use of regorafenib for patients with mCRC who are refractory to chemotherapy; it is to be given before or after FTD/TPI, FTD/TPI plus bevacizumab, or fruquintinib. They also recommend use of the dose-escalation strategy tested in the ReDOS trial as an appropriate alternative dosing approach.2 Patients receiving regorafenib may experience AEs such as fatigue, HFSR, and hypertension; still, this medication may be an option in patients for whom a chemotherapy-sparing option is needed and AEs such as myelosuppression must be avoided.
Trifluridine/tipiracil
FTD/TPI, an oral chemotherapeutic agent, was approved by the FDA in 2015 for the treatment of adults with mCRC who have previously received fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy; an anti-VEGF biologic therapy; and, if RAS wild-type, an anti-EGFR therapy.35 FTD/TPI consists of 2 agents: trifluridine (a thymidine-based nucleoside analog) and tipiracil (a thymidine phosphorylase inhibitor that helps prevent the breakdown of active trifluridine). Trifluridine is incorporated into the DNA of cancer cells, disrupting the process of DNA synthesis and hindering cellular growth.35
In the RECOURSE clinical trial (NCT01607957), investigators evaluated FTD/TPI monotherapy.36 In this randomized, double-blind, placebo-controlled trial, 534 patients with mCRC who were previously treated with at least 2 lines of chemotherapy were randomly assigned to receive FTD/TPI plus BSC, and 266 participants were givenplacebo plus BSC. FTD/TPI was taken orally twice daily on days 1 to 5 and 8 to 12 of a 28-day treatment cycle. The primary end point evaluated was OS, and PFS was a secondary end point.
Treatment with FTD/TPI demonstrated statistically significant improvements in both OS and PFS as compared with placebo. The median OS among patients receiving FTD/TPI was 7.1 months compared with 5.3 months for those receiving placebo (HR, 0.68; 95% CI, 0.58-0.81; P < .001).36 Among patients treated in the United States, median PFS was 2.0 vs 1.7 months, respectively (HR, 0.48; 95% CI, 0.41-0.57; P < .001).
In the RECOURSE trial, 53% of patients treated with FTD/TPI required a delay of at least 4 days before beginning the next cycle of treatment due to toxicity, 14% of patients required a dose reduction, and 4% of patients discontinued treatment due to AEs.36 AEs of at least grade 3 were observed in 69% of patients treated with FTD/TPI; neutropenia, anemia, febrile neutropenia, fatigue, and diarrhea were the most common AEs or laboratory anomalies leading to dose reduction.
Trifluridine/tipiracil With Bevacizumab
Subsequent to the RECOURSE trial, FTD/TPI was evaluated as combination therapy with bevacizumab in the SUNLIGHT trial (NCT04737187); results of this study ultimately led to FDA approval of this combination in August 2023.35 This international, randomized, active-controlled, open-label study involved patients previously treated with mCRC and required that patients have no more than 2 prior treatments for mCRC.17 In all, 246 patients were randomly assigned to receive FTD/TPI plus bevacizumab, and 246 patients were given FTD/TPI alone. FTD/TPI was administered by mouth twice daily on days 1 to 5 and 8 to 12 of a 28-day treatment cycle, with bevacizumab administered intravenously on days 1 and 15 of each 4-week treatment cycle. The primary treatment end point evaluated was OS, with PFS being a secondary outcome measure.
FTD/TPI plus bevacizumab proved to be statistically superior to FTD/TPI monotherapy for both OS and PFS.17 Patients receiving FTD/TPI plus bevacizumab demonstrated a median OS of 10.8 months compared with 7.5 months for those receiving FTD/TPI alone (HR, 0.61; 95% CI, 0.49-0.77; P < .001). The median PFS was 5.6 vs 2.4 months, respectively (HR, 0.44; 95% CI, 0.36-0.54; P < .001). Notably, survival benefits were significantly better with combination treatment regardless of whether patients received previous bevacizumab therapy. However, while patients benefited from treatment with FTD/TPI plus bevacizumab regardless of prior bevacizumab exposure, the benefit was meaningfully larger in patients who had not been exposed to prior bevacizumab. In a subgroup analysis, the median PFS was 7.7 months (95% CI, 6.2-9.4 months) in patients who did not receive prior bevacizumab but 4.5 months (95% CI, 4.1-5.5 months) in those given prior bevacizumab; the median OS was 15.1 months with prior bevacizumab (95% CI, 12.1 months to not evaluable) but 9.0 months without (95% CI, 8.3-10.8 months); however, neither improvement was statistically significant.37
Severe AEs (grade ≥ 3) were reported in 72.4% of patients in the FTD/TPI plus bevacizumab group and 69.5% of the FTD/TPI monotherapy group.17 The most common AEs (any grade) in both groups were neutropenia, nausea, and anemia. Both the combination and monotherapy groups experienced hematologic toxicity of at least grade 3; specifically, neutropenia (43.1% and 32.1%, respectively), anemia (6.1% and 11.0%, respectively), and thrombocytopenia (2.8% and 1.2%, respectively) were noted. Granulocyte colony stimulating factors were given to 29.3% and 19.5% of patients in the FTD/TPI plus bevacizumab group and FTD/TPI monotherapy group, respectively.17 Dose delays were common, occurring in 69.5% of patients using combination therapy and 53.3% of those given monotherapy; AE-related treatment discontinuation occurred in 12.6% of patients in both groups.
FTD/TPI alone or with bevacizumab is recommended by the NCCN Guidelines as an option for patients experiencing disease progression despite standard treatments. The NCCN Guidelines offer no specific guidance or recommendation about sequencing therapy between FTD/TPI, regorafenib, or fruquintinib for patients in the 3L+ setting; however, it does recommend use of FTD/TPI plus bevacizumab over FTD/TPI monotherapy.2 FTD/TPI given alone or with bevacizumab may be an option for patients needing to avoid toxicities such as HFSR, but its use may be challenging for those who are unable to tolerate myelosuppression.
Fruquintinib
Fruquintinib, an oral kinase inhibitor approved by the FDA in October 2023, is indicated for patients with mCRC who were previously treated with chemotherapy and biologic agents, having received all of the following: fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy; an anti-VEGF therapy; and an anti-EGFR therapy if medically appropriate.38 Fruquintinib is a highly selective, small-molecule VEGF inhibitor that selectively inhibits VEGFR1, -2, and -3 to slow tumor growth while having a weak to no inhibitory effect on other kinases.39
Efficacy of fruquintinib was evaluated in the FRESCO-2 trial (NCT04322539), an international, multicenter, randomized, double-blind, placebo-controlled study that randomly assigned 691 patients with mCRC who had disease progression in a 2:1 ratio; 461 patients received fruquintinib orally once daily for the first 21 days of a 28-day cycle plus BSC, and 230 patients received placebo plus BSC.40 Notably, a majority of patients (73%) had more than 3 lines of previous therapy (median, 4 previous lines of therapy). Additionally, all patients had previously received regorafenib, FTD/TPI, or both agents. Efficacy was evaluated using the primary end point of OS and the secondary end point of PFS.
Adding fruquintinib to BSC resulted in a median OS of 7.4 months compared with 4.8 months with use of placebo plus BSC (HR, 0.66; 95% CI, 0.55-0.80; P < .001).40 Fruquintinib plus BSC also demonstrated significant improvements in PFS (3.7 vs 1.8 months, respectively; HR, 0.32; 95% CI, 0.27-0.39; P < .001).
Grade 3 or worse AEs occurred in 63% of patients in the fruquintinib group, the most frequent being hypertension (14%), asthenia (8%), and HFSR (6%).40 One treatment-related death due to intestinal perforation was reported in the fruquintinib group. AEs in patients receiving fruquintinib resulted in 20% of patients discontinuing treatment, 47% experiencing dose interruptions, and 24% having dose reductions. Among 110 patients, the need to reduce the dose was most frequently attributed to HFSR (24 patients), hypertension (17 patients), and asthenia (16 patients).40 Of note, no hematologic toxicity was observed with fruquintinib.
The NCCN Guidelines include fruquintinib as a recommended therapy for patients with previously treated mCRC who experienced progression with use of all available regimens.2 Similar to those given regorafenib, patients receiving fruquintinib may experience AEs such as fatigue, HFSR, and hypertension. However, fruquintinib therapy is another option when chemotherapy (and subsequent myelosuppression) is being avoided.
Sequencing Considerations in the Third Line and Later
To date, no head-to-head or sequencing studies have been conducted to compare these therapies.41 NCCN Guidelines recognize and recommend regorafenib, FTD/TPI, FTD/TPI plus bevacizumab, and fruquintinib for later-line treatment in patients with refractory disease; however, they acknowledge that no data inform the best order of those therapies, and they state that any of the therapies may be given before or after the others with preference for FTD/TPI plus bevacizumab over FTD/TPI monotherapy.2 The complexity of this decision highlights the importance of ensuring availability of all 4 therapies to patients with mCRC so that optimal treatment decisions can be made on a patient-specific basis.
Treating with regorafenib prior to FTD/TPI in the 3L may be preferred due to several factors.42 First, many patients reaching 3L treatment may have experienced hematologic AEs from exposure to multiple lines of chemotherapy. A chemotherapy-free interval (ie an interval in which alternative therapies such as targeted therapies or immunotherapy are used) may prove beneficial for select patients whose bone marrow reserves are depleted with use of previous chemotherapy regimens. Additionally, chemotherapy given after regorafenib appears to maintain clinical benefit, and regorafenib may offer a different mechanism of action at this stage of the disease.42 Similar to regorafenib, fruquintinib offers a mechanism of action that differs from more conventional chemotherapy to use in the 3L setting and would offer a chemotherapy-free interval with low risk of hematologic toxicity.38
Unmet Needs in Third-Line Treatments
Treatment of patients with cytotoxic therapies in earlier lines can result in hematologic AEs, including anemia and neutropenia. Patients who experience myelosuppression during earlier cycles of chemotherapy may be at increased risk for neutropenic complications in subsequent cycles.43 Long-term use of chemotherapy is often associated with myelosuppression and other hematologic AEs, highlighting the unmet need for other therapeutic strategies.29 Prolonging life while maintaining patient QOL are key treatment objectives for patients with advanced mCRC.44 With the overarching strategy of preserving QOL to the highest degree possible, it’s important that patients be exposed to all viable therapeutic options.44
Additional studies are needed to determine optimal sequencing of therapies in the 3L and beyond with options such as FTD/TPI alone or with bevacizumab, regorafenib, and fruquintinib to best navigate potential toxicities, preserve patient QOL, and improve OS. More research in the area of relevant and targetable biomarkers is needed in this space to further advance our precision medicine approaches in mCRC.
Conclusion
There has been substantial progress in the treatment of mCRC with ongoing research about the role of predictive biomarkers, creating a complex and evolving clinical landscape. Most patients with mCRC who become refractory to initial lines of chemotherapy become eligible for additional lines of treatment with the goal of controlling disease and preserving an acceptable QOL. Importantly, as the number of lines of treatment increases, the likelihood of treatment-limiting toxicities may also increase. Therapies such as regorafenib, FTD/TPI alone or with bevacizumab, and fruquintinib are recommended by the NCCN Guidelines for use in this setting with treatment decisions relying heavily on patient-specific characteristics. Clinical pathways should retain flexibility to allow therapeutic selection based on patient-specific situations. Details about prior therapy, a patient’s individual experience of treatment-related toxicities, and a nuanced understanding of a patient’s tumor characteristics should all be weighed carefully when selecting treatment for mCRC in the 3L and beyond.
Authorship Affiliation: Department of Hematology and Medical Oncology, Mayo Clinic, Phoenix, AZ
Source of Funding: This supplement was supported by Bayer US, LLC.
Author Disclosures: Dr Bekaii-Saab reports the following personal conflicts of interest: serving as a consultant to Abbvie, Inc; Aptitude Health; AstraZeneca; BeiGene, Inc; Blueprint Medicines Corporation; Boehringer Ingelheim Pharmaceuticals, Inc; Celularity Inc; Daiichi Sankyo Company Limited; Deciphera Pharmaceuticals, Inc; Exact Sciences Corporation, Exelixis, Inc; Foundation Medicine, Inc; GSK plc; Illumina, Inc; Janssen Pharmaceuticals, Inc; Kanaph Therapeutics Inc; Lisata Therapeutics, Inc; MJH Life Sciences; Natera, Inc; Sanofi; Swedish Orphan Biovitrum AB; Stemline Therapeutics, Inc; Treos Bio Limited; Xilio Therapeutics; and Zai Lab; serving as a scientific advisory board member for Artiva Biotherapeutics, Inc; AstraZeneca; Eisai Co, Ltd; Exelixis, Inc; FibroGen, Inc; Immuneering Corporation; Imugene Limited; Kintor Pharmaceutical Limited; Merck & Co, Inc; Pancreatic Cancer Action Network; Replimune Group Inc; Sun Pharmaceutical Industries Ltd; Valley Health System; Xilis; and 1Globe Health; and receiving royalties from UpToDate. He also reports an additional personal conflict of interest: inventions/patents 18/183,488: Human PD1 peptide vaccines and uses thereof, 19/055,687: Methods and compositions for the treatment of cancer cachexia. He reports the following institutional conflicts of interest: receiving research funding from Agios Pharmaceuticals, Inc; Arcus Biosciences, Inc; Arys Medical; Atreca, Inc; Bayer US, LLC; Boston Biomedical, Inc; Bristol Myers Squibb Company; Celgene Corporation; Clovis Oncology, Inc; Eisai Co, Ltd; Genentech, Inc; Incyte; Ipsen Pharma; Lilly; Merus; Mirati Therapeutics, Inc; Novartis AG; Pfizer Inc; and Seagen Inc, and serving as a consultant to Arcus Biosciences, Inc; Bayer US, LLC; Eisai Co, Ltd; Genentech, Inc; Incyte; Ipsen Pharma; Les Laboratoires Servier; Merck & Co, Inc; Merck KGaA; Merus; Pfizer Inc; and Seagen Inc.
Authorship Information: Concept and design; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content.
Address Correspondence To: Tanios Bekaii-Saab, MD. Mayo Clinic, 5881 East Mayo Boulevard, Phoenix, AZ, 85054. Email: bekaii-saab.tanios@mayo.edu
REFERENCES
- Cancer Facts & Figures 2023. American Cancer Society; 2023. Accessed November 21, 2023. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2023/2023-cancer-facts-and-figures.pdf
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Colon Cancer. V.1.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed January 30, 2024. To view the most recent and complete version of the guideline, go online to NCCN.org.
- Cancer stat facts: colorectal cancer. National Cancer Institute. Updated August 2023. Accessed November 14, 2023. https://seer.cancer.gov/statfacts/html/colorect.html
- Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325(7):669-685. doi:10.1001/jama.2021.0106
- Financial burden of cancer care. National Cancer Institute. August 2023. Accessed November 14, 2023. https://progressreport.cancer.gov/after/economic_burden
- Bhimani N, Wong GYM, Molloy C, et al. Cost of treating metastatic colorectal cancer: a systematic review. Public Health. 2022;211:97-104. doi:10.1016/j.puhe.2022.06.022
- Latremouille-Viau D, Chang J, Guerin A, et al. The economic burden of common adverse events associated with metastatic colorectal cancer treatment in the United States. J Med Econ. 2017;20(1):54-62.
doi:10.1080/13696998.2016.122557 - Harada S, Morlote D. Molecular pathology of colorectal cancer. Adv Anat Pathol. 2020;27(1):20-26. doi:10.1097/PAP.0000000000000247
- Malki A, ElRuz RA, Gupta I, Allouch A, Vranic S, Al Moustafa AE. Molecular mechanisms of colon cancer progression and metastasis: recent insights and advancements. Int J Mol Sci. 2020;22(1):130. doi:10.3390/ijms22010130
- Prior IA, Hood FE, Hartley JL. The frequency of RAS mutations in cancer. Cancer Res. 2020;80(14):2969-2974. doi:0.1158/0008-5472.CAN-19-3682
- Yoshino T, Di Bartolomeo M, Raghav K, et al; DESTINY-CRC01 Investigators. Final results of DESTINY-CRC01 investigating trastuzumab deruxtecan in patients with HER2-expressing metastatic colorectal cancer. Nat Commun. 2023; 14(1):3332. doi:10.1038/s41467-023-38032-4
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Rectal Cancer V.1.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed January 30, 2024. To view the most recent and complete version of the guideline, go online to NCCN.org.
- Sun W. Angiogenesis in metastatic colorectal cancer and the benefits of targeted therapy. J Hematol Oncol. 2012;5:63. doi:10.1186/1756-8722-5-63
- Avastin. Prescribing information. Genentech; 2022. Accessed February 14, 2024.
https://www.gene.com/download/pdf/avastin_prescribing.pdf - Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335-2342. doi:10.1056/NEJMoa032691
- Bennouna J, Sastre J, Arnold D, et al; ML18147 Study Investigators. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14(1):29-37. doi:10.1016/S1470-2045(12)70477-1
- Prager GW, Taieb J, Fakih M, et al; SUNLIGHT Investigators. Trifluridine-tipiracil and bevacizumab in refractory metastatic colorectal cancer. N Engl J Med. 2023;388(18):1657-1667. doi:10.1056/NEJMoa2214963
- Erbitux. Prescribing information. ImClone/Lilly; 2021. Accessed February 14, 2024. https://uspl.lilly.com/erbitux/erbitux.html#pi
- Vectibix. Prescribing information. Amgen; 2021. Accessed February 14, 2024. https://www.pi.amgen.com/-/media/Project/Amgen/Repository/pi-amgen-com/vectibix/vectibix_pi.pdf
- Zhao B, Wang L, Qiu H, et al. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget. 2017;8(3):3980-4000. doi:10.18632/oncotarget.14012
- Bekaii-Saab TS, Lach K, Hsu LI, et al. Impact of anti-EGFR therapies on HER2-positive metastatic colorectal cancer: a systematic literature review and meta-analysis of clinical outcomes. Oncologist. 2023;28(10):885-893. doi:10.1093/oncolo/oyad200
- Rossini D, Boccaccino A, Carullo M, et al. Primary tumour side as a driver for treatment choice in RAS wild-type metastatic colorectal cancer patients: a systematic review and pooled analysis of randomised trials. Eur J Cancer. 2023;184:106-116. doi:10.1016/j.ejca.2023.02.006
- Keytruda. Prescribing information. Merck & Co; 2024. Accessed February 14, 2024.
https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf - Opdivo. Prescribing information. Bristol Myers Squibb; 2023. Accessed February 14, 2024.
https://packageinserts.bms.com/pi/pi_opdivo.pdf - Van Cutsem E, Cervantes A, Adam R, et al; RECOURSE Study Group. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386-1422. doi:10.1093/annonc/mdw235
- Bekaii-Saab T, Kim R, Kim TW, et al. Third- or later-line therapy for metastatic colorectal cancer: reviewing best practice. Clin Colorectal Cancer. 2019;18(1):e117-e129. doi:10.1016/j.clcc.2018.11.002
- Fernández-Montes A, Grávalos C, Pericay C, et al. Current options for third-line and beyond treatment of metastatic colorectal cancer. Spanish TTD Group Expert Opinion. Clin Colorectal Cancer. 2020;19(3):165-177. doi:10.1016/j.clcc.2020.04.003
- Byrne M, Saif MW. Selecting treatment options in refractory metastatic colorectal cancer. Onco Targets Ther. 2019;12:2271-2278. doi:10.2147/OTT.S194605
- Epstein RS, Aapro MS, Basu Roy UK, et al. Patient burden and real-world management of chemotherapy-induced myelosuppression: results from an online survey of patients with solid tumors. Adv Ther. 2020;37(8):3606-3618. doi:10.1007/s12325-020-01419-6
- Stivarga. Prescribing information. Bayer HealthCare Pharmaceuticals; 2020. Accessed February 14, 2024. https://labeling.bayerhealthcare.com/html/products/pi/Stivarga_PI.pdf
- Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303-312. doi:10.1016/S0140-6736(12)61900-X
- Li J, Qin S, Xu R, et al; CONCUR Investigators. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015;16(6):619-629. doi:10.1016/S1470-2045(15)70156-7
- Bekaii-Saab TS, Ou FS, Ahn DH, et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): a randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019;20(8):1070-1082. doi:10.1016/S1470-2045(19)30272-4
- Argiles G, Mulet N, Valladares-Ayerbes M, et al; Spanish Cooperative Group for the Treatment of Digestive Tumors (TTD) and UNICANCER GI; REARRANGE Investigators and Steering Committee. A randomised phase 2 study comparing different dose approaches of induction treatment of regorafenib in previously treated metastatic colorectal cancer patients (REARRANGE trial). Eur J Cancer. 2022;177:154-163. doi:10.1016/j.ejca.2022.09.037
- Lonsurf. Prescribing information. Taiho Oncology; 2023. Accessed February 14, 2024.
https://taihocorp-media-release.s3.us-west-2.amazonaws.com/documents/prescribing-information.pdf - Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372(20):1909-1919. doi:10.1056/NEJMoa1414325
- Prager GW, Taieb J, Fakih M, et al; SUNLIGHT Investigators. Trifluridine-tipiracil and bevacizumab in refractory metastatic colorectal cancer. Supplementary appendix. N Engl J Med. 2023;388(18):1657-1667. doi:10.1056/NEJMoa2214963
- Fruzaqla. Prescribing information. Takeda Pharmaceuticals; 2023. Accessed February 14, 2024. https://www.fruzaqla.com/sites/default/files/resources/fruzaqla-prescribing-information.pdf
- Sun Q, Zhou J, Zhang Z, et al. Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol Ther. 2014;15(12):1635-1645. doi:10.4161/15384047.2014.964087
- Dasari A, Lonardi S, Garcia-Carbonero R, et al; FRESCO-2 Study Investigators. Fruquintinib versus placebo in patients with refractory metastatic colorectal cancer (FRESCO-2): an international, multicentre, randomised, double-blind, phase 3 study. Lancet. 2023;402(10395):41-53. doi:10.1016/S0140-6736(23)00772-9
- Tabernero J, Taieb J, Prager GW, et al. Trifluridine/tipiracil plus bevacizumab for third-line management of metastatic colorectal cancer: SUNLIGHT study design. Future Oncol. 2021;17(16):1977-1985. doi:10.2217/fon-2020-1238
- Grothey A, Ciardiello F, Marshall JL. How to incorporate a chemo-free interval into the management of metastatic colorectal cancer. Clin Adv Hematol Oncol. 2020;18(10; suppl 16):1-24.
- Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia: risks, consequences, and new directions for its management. Cancer. 2004;100(2):228-237. doi:10.1002/cncr.11882
- Riedesser JE, Ebert MP, Betge J. Precision medicine for metastatic colorectal cancer in clinical practice. Ther Adv Med Oncol. 2022;14:1-25. doi:10.1177/17588359211072703

